



来源:《临床儿科杂志》
赵培伟, 张蕾, 孟庆杰, 何学莲. 脑组织铁沉积神经变性病患儿的临床与遗传学分析 [J]. 临床儿科杂志, 2025, 43(3): 199-203 DOI:10.12372/jcp.2025.24e0318
ZHAO Peiwei, ZHANG Lei, MENG Qingjie, HE Xuelian. Clinical and genetic features of seven patients with neurodegeneration with brain iron accumulation[J]. Journal of Clinical Pediatrics, 2025, 43(3): 199-203 DOI:10.12372/jcp.2025.24e0318
本文作者:赵培伟 1 张 蕾 1 孟庆杰 2 何学莲 1
作者单位:华中科技大学同济医学院附属武汉儿童医院 1. 精准医学实验室,2. 检验科(湖北武汉 430016)
摘要:目的 探讨 7 例脑组织铁沉积神经变性病(NBIA)患儿的临床及遗传学特点。方法 利用全外显子测序技术(WES)对疑似 NBIA 的患儿进行遗传学检测,对 WES 检测阳性的患儿通过 Sanger 测序验证;回顾性分析患儿临床资料并进行文献复习。结果 本研究纳入 7 例患儿,患儿首发症状为步态障碍、姿势异常、精神运动发育迟滞或倒退等。WES 检测发现 7 例患儿均分别携带 NBIA 相关的致病基因,其中 PANK2 基因 1 例、FA2H 基因 1 例以及 PLA2G6 基因 5 例。PANK2 基因异常的患儿临床表现为步态障碍及姿势异常、视网膜色素变性-苍白球变性综合征等典型的症状;FA2H 异常的患儿表现为语言落后、步态异常以及肌张力障碍;PLA2G6 异常的患儿起病较早,表现为发育落后、肌无力、苍白球低信号铁沉积及小脑萎缩。结论 NBIA 患儿临床表型复杂且不同类型 NBIA 患儿临床表现存在差异,神经系统异常为最主要的临床表现。
关键词:脑组织铁沉积神经变性病;全外显子测序;临床表型;儿童
脑组织铁沉积神经变性病 (neurodegeneration with brain iron accumulation,NBIA) 是一组由基因变异导致的以锥体外系症状为主,伴有其他复杂临床症状,在脑组织特定部位可见异常铁沉积的罕见的神经遗传变性疾病;发病率约 1~3/百万 [1-2]。NBIA 有多种临床亚型,目前文献及数据库报道 NBIA 疾病谱系中 10 个亚型已经明确了致病基因,分别为 PANK2、PLA2G6、WDR45、C19orf12、FA2H、CP、FTL、ATP13A2、DCAF17 以及 COASY;其中 WDR45 与 FTL 为显性遗传,其余基因均为常染色体隐性遗传 [3-5]。其中患儿中最常见的为 PANK2 基因变异所导致 Hallervorden-Spatz 病(约占 50%)[6],其他的常见类型是非钙依赖型磷脂酶 A2 相关性神经变性病(phospholipase A2 associated neurodegeneration,PLAN,约占 20%)、线粒体膜蛋白相关性神经变性病(mitoehondrial membrane protein associated neuro-degeneration,MPAN,约占 10%)等;但仍有 10% 的 NBIA 病例目前尚未明确致病基因 [7]。本研究分析了 7 例 NBIA 患儿,探讨该疾病的临床表现以及遗传学特点。
1 对象与方法
1.1 研究对象
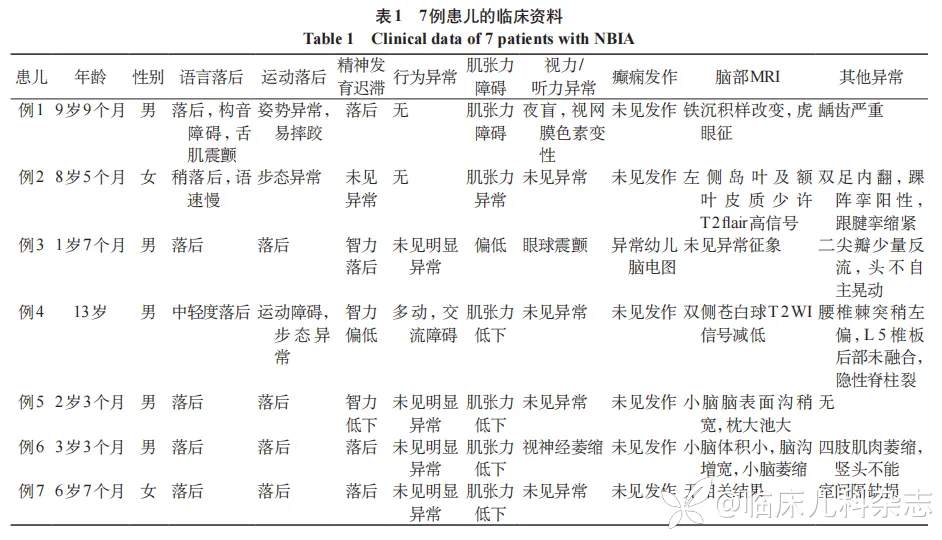
回顾性分析 2018 年 6 月至 2022 年 6 月在武汉儿童医院就诊,首发症状多为肌张力异常、步态障碍及姿势异常、精神运动发育迟滞或倒退的 23 例患儿,经基因检测明确诊断为 NBIA 的患儿 7 例。依据临床表现,患儿初步诊断为发育迟滞或肌张力障碍等。本研究得到医院伦理委员会的审批(No.2020R006-E05)以及得到患儿家属的知情同意。收集 7 例患儿的临床资料,包括现病史、家族史、发病年龄、体格检查、实验室检查、以及听力视力筛查、脑电图检查、头颅 MRI 等辅助检查结果(见表 1)。
1.2 全外显子测序分析
采集患儿及其父母外周静脉血 2 mL,采用北京天根外周血基因组 DNA 提取试剂盒提取患儿 DNA 并测定浓度,由第三方检测公司(北京智因东方转化医学有限公司)进行全外显子测序,测序的原始数据与参考基因组(GRCh38 版本)进行比对,然后采用 GATK 以及 VarScan 软件进行变异注释分析,利用 Annovar 软件及数据库评定变异位点的生物学影响;候选致病基因采用 Sanger 测序技术验证患儿及家属的变异位点,并利用美国医学遗传学与基因组学学会(ACMG)指南评估变异的临床意义。
2 结果
2.1 患儿基本信息
7 例患儿,其中 5 例男性,2 例女性,平均年龄(5.4±3.8)岁;患儿在产前均未见明显异常,出生以后面容未见异常;患儿均存在语言及运动落后的表现,且患儿均存在不同程度的肌张力障碍,临床表型见表 1。
2.2 辅助检查
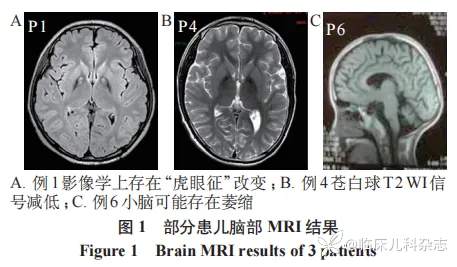
2 例患儿(例 1 和例 6)存在视神经萎缩,患儿例 3 有眼球震颤的表型。7 例患儿中有 5 例患儿脑部 MRI 结果存在异常,其中患儿例 1 存在典型的虎眼征样改变;其他患儿存在因脑部部分区域铁沉积导致的 T2 flair 信号改变或者小脑萎缩,见图 1。
2.3 遗传学分析
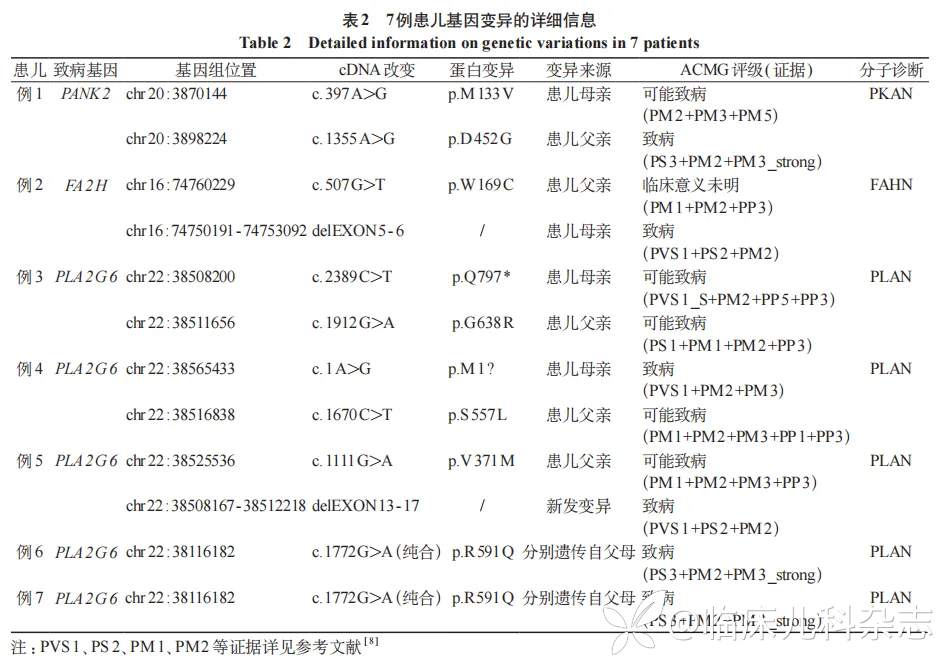
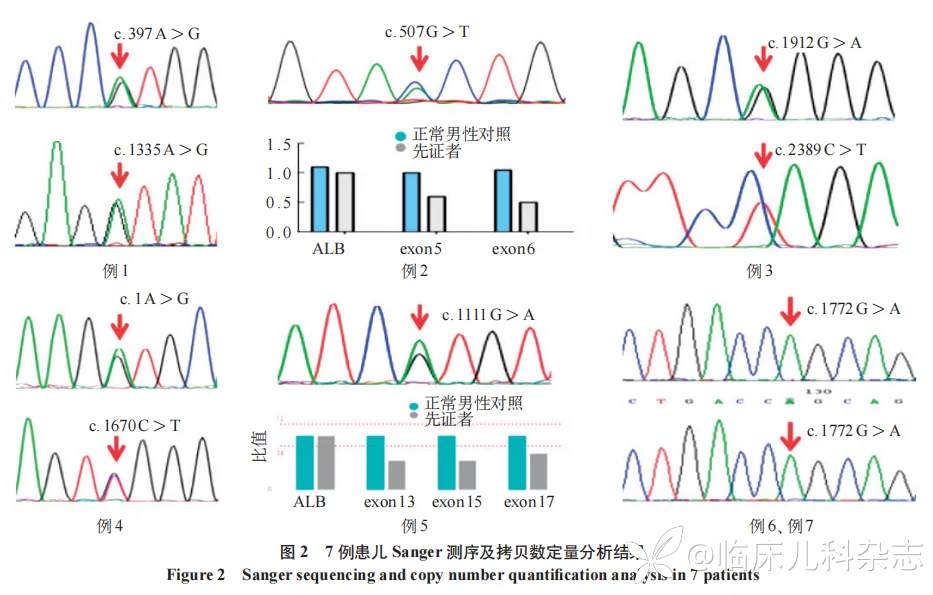
高通量测序产生的原始数据经过生物信息学软件分析注释以后,发现 7 例患儿均存在致病基因变异,经过 Sanger 测序验证发现例 1 存在 PANK2 基因复合杂合变异(c.397A>G 和 c.1335A>G);例 2 存在 FA2H 基因复合杂合变异(c.507 G>T 和 del EXON5-6)。例 3~例 7 均携带 PLA2G6 基因的变异,其中例 6 与例 7 是兄妹关系,两人均携带 PLA2G6 基因的 c.1772 G>A 纯合变异;依据 ACMG 指南以上变异均为致病或可能致病性改变,变异详细信息见表 2、图 2;其中 FA2H 基因外显子 5-6 缺失以及 PLA2G6 基因外显子 13-17 缺失未见报道。
3 讨论
脑组织铁沉积神经变性病(NBIA)是一组罕见的神经遗传变性疾病;本研究报道 7 例经高通量测序确诊的 NBIA 患儿,其中携带 PANK2 基因变异 1 例、FA2H 基因 1 例以及 PLA2G6 基因变异 5 例。基因检测协助临床及时明确诊断并发现两个未见报道的新变异。
PANK2 基因变异导致泛酸激酶相关性神经变性病(PKAN),该疾病也称为苍白球黑质红核色素变性或 Hallervorden-Spatz 病,为常染色隐性遗传;该类疾病约占 NBIA 患儿的 50%[4,8]。PANK2 定位于染色体 20p13,长约 1.85kb。PANK2 在辅酶 A 的生物合成中起关键作用。根据发病年龄,PKAN 可分为早发典型 PKAN 和晚发不典型 PKAN[9]。本研究中 1 例患儿 3 岁以内起病,临床表现为舌肌震颤,右侧面瘫;行走姿势异常,肌张力障碍,视网膜色素变性,学习成绩下降,轻到中度智力低下,脑部 MRI 为虎眼征,患儿为典型的 PKAN。目前临床对该类疾病的治疗主要是对症及预防并发症治疗;后期随访发现本例患儿口服硝西泮、苯海索片等治疗。另外国际上又对数十例 PKAN 患儿施行脑深部电刺激手术(deep brain stimulation,DBS)[10],长期运动症状的改善率仅为 30%~50%,确切的疗效需要进一步的验证。
PLA2G6 基因变异导致非钙依赖型磷脂酶 A2 相关性神经变性病(PLAN),为常染色体隐性遗传 [4]。PLA2G6 位于染色体 22q13,有 17 个外显子,编码非钙依赖型磷脂酶 A2-β蛋白,参与细胞膜磷脂的转换、线粒体功能调节以及铁离子的代谢等 [11]。PLAN 可导致 3 种疾病:婴儿神经轴索营养不良(infantile neuroaxonal dystrophy,INAD)、脑组织铁沉积神经变性病 2B 型以及 PLA2G6 相关性肌张力障碍-帕金森综合征(PLA2G6-associated dystonia-parkinsonism,PLAN-DP)[12]。典型 INAD 病例中,小脑蚓部和小脑半球的萎缩是最主要和特征性的影像学表现。约有 50%INAD 病例可出现异常铁沉积,部分患儿存在胼胝体压部萎缩变细的表现 [13]。本研究的纳入 5 例 PLA2G6 基因变异的患儿,除例 4 外,其余病例均起病较早(6 个月~3 岁以内),有 2 例患儿存在小脑萎缩或小脑体积减少/脑沟增宽的现象。PLAN 具有明显的临床异质性,导致临床上对于锥体外系症状患儿的诊断十分困难,容易导致患儿的误诊;研究发现基因型和表型存在一定相关性:变异位于 PLA2G6 基因外显子 7 和 16 上的患儿与 PLAN-DP 存在相关性 [13]。本类疾病患儿多予对症治疗,对帕金森样症状,左旋多巴治疗可能有效;具有 INAD 和非典型 NAD 患儿可口服或注射巴氯芬治疗肌张力障碍 [14]。
FA2H 基因变异导致脂肪酸羟化酶相关性神经变性病(FAHN)。FA2H 蛋白质能催化 2-羟基化脂肪酸,基因变异可以导致髓鞘生成障碍 [15]。研究发现 FA2H 基因变异可以导致脑白质营养不良、遗传性痉挛性截瘫 35 型(HSP35)[16],部分患儿脑组织内有异常铁沉积,因此将其归类于 NBIA 的一个亚型,称为 FAHN。患儿常见的表型为步态障碍、容易跌倒,逐渐进展为痉挛性步态、肌张力障碍、小脑性共济失调、构音障碍、吞咽障碍、视神经萎缩致视力障碍等 [17-18]。本研究中的例 2 临床症状较为典型,临床初步诊断为遗传性痉挛性截瘫,结合分子检测结果,患儿可诊断为 FAHN,与已报道的患儿的表型相似。和其他类型的 NBIA 一样,FAHN 治疗还缺乏有效的手段,目前仍然是对症治疗、营养支持、预防并发症等;但有研究表明脐带间充质干细胞移植治疗可能对延缓疾病进展有一定的疗效。
综上所述,脑组织铁沉积性神经变性疾病是一类由特定基因变异所致的神经系统遗传性疾病,临床主要表现为运动障碍、步态异常、痉挛、认知障碍等,其共同的影像学特征是脑内特定部位出现异常铁离子沉积信号。患儿临床异质性较高,全外显子测序有助于临床早期明确诊断;本研究发现两个未见报道的缺失变异,增加了该类疾病的变异谱。
好文章,需要你的鼓励