



来源:临床儿科杂志
作者:黄海生 1,2 赵安琪 2 曾 琴 2 王雨蒙 2 何 伟 2 李 明 1,2
作者单位:1. 安徽理工大学第一附属医院;2. 复旦大学附属儿童医院皮肤科
基金项目:国家自然科学基金资助项目
通讯作者:李明 电子邮箱:mingli@fudan.edu.cn
推荐引用格式:
黄海生, 赵安琪, 曾琴, 等.PSAT1 基因变异致轻型丝氨酸缺乏症:以鱼鳞病合并周围神经病为表型的临床特征与遗传学分析 [J]. 临床儿科杂志, 2026, 44(5): 424-430 DOI:10.12372/ jcp.2026.25e1598
HUANG Haisheng, ZHAO Anqi, ZENG Qin, et al.PSAT1 gene variant causes mild serine deficiency: clinical and genetic analysis of ichthyosis combined with peripheral neuropathy[J].Journal of Clinical Pediatrics, 2026, 44(5): 424-430 DOI:10.12372/jcp.2026.25e1598
摘要
目的 探讨 PSAT1 基因变异丝氨酸缺乏症的临床分型、遗传学基础及诊疗策略,提升临床对该病的识别能力。方法 回顾性收集 1 例以鱼鳞病合并周围神经病为表现的患儿临床资料,通过家系全外显子组测序、Sanger 验证及蛋白质结构预测研究其遗传学病理机制,并随访其治疗结果。系统检索数据进行文献复习,总结其临床表型、基因变异特征、治疗反应及预后特点。结果 患儿女,12 岁,2 岁起出现全身皮肤干燥脱屑,7 岁起进行性双下肢无力、足下垂,伴水平眼震。既往检查示血浆丝氨酸水平正常(233.29μmol/L、319.19μmol/L),家系全外显子组测序检出 PSAT1 基因新纯合变异 c.697C>A(p.Q233K),ACMG 评级为意义未明变异,Sanger 验证显示父母均为携带者。予以患者口服 L-丝氨酸(300 mg·kg-1·d-1)和甘氨酸(200 mg·kg-1·d-1)补充治疗,1 周后皮肤干燥脱屑明显好转,13 周后行走步态显著改善,可自主下蹲站起。文献复习共纳入 5 例轻型丝氨酸缺乏症,结合本例共 6 例,均为 PSAT1 基因变异。结论 丝氨酸缺乏症表型谱广泛,轻型以儿童期鱼鳞病合并青少年期周围神经病为核心表现,因血浆丝氨酸水平多正常或轻度降低极易漏诊。早期诊断与及时补充 L-丝氨酸对改善预后至关重要,可避免不可逆神经损伤。对原因不明的鱼鳞病伴周围神经病变患者,应尽早行基因检测以明确诊断。
关键词
PSAT1 基因;轻型丝氨酸缺乏症;鱼鳞病合并周围神经病
引言
丝氨酸缺乏症是一组由 L-丝氨酸生物合成通路中三种合成酶之一发生缺陷所引起的常染色体隐性遗传性代谢疾病 [1]。L-丝氨酸作为非必需氨基酸,其生物合成依赖于三种关键酶构成的酶系统,包括由 PHGDH 基因编码的磷酸甘油酸脱氢酶(phosphoglycerate dehydrogenase,PGDH)、由 PSAT1 基因编码的磷酸丝氨酸氨基转移酶(phosphoserine aminotransferase,PSAT)以及由 PSPH 基因编码的磷酸丝氨酸磷酸酶(phosphoserine phosphatase,PSP)[2]。丝氨酸缺乏症表型谱广泛,根据临床表型的严重程度可分为致死型、非致死型和轻型。其中以儿童期鱼鳞病和青少年期周围神经病为主要表现的轻型丝氨酸缺乏症罕见,目前国内外仅有 5 例报道 [2-4]。本文报道 1 例 PSAT1 基因变异导致的轻型丝氨酸缺乏症并给予治疗,探讨其临床与遗传学特点,并对既往文献中的相关病例进行总结。
1 对象与方法
1.1 研究对象
患儿,女,12 岁以「全身皮肤干燥 10 年余,双下肢行走困难 5 年余」为主诉于 2025 年 7 月就诊复旦大学附属儿科医院皮肤科。
1.2 方法
1.2.1 临床资料 收集患儿临床资料,包括性别、年龄、起病年龄、现病史、个人史、家族史等病史资料,由专业的皮肤科医师对患者进行系统体格检查。同时,对患儿进行肌电图检测、肌肉活检,以及脑脊液与血清周围神经病变抗体检测。
1.2.2 遗传学检测 在获得知情同意后采集患儿及父母抗凝血用磁珠法血液基因组提取试剂盒(天根生化,中国)提取基因组 DNA 并进行质检;采用 xGen® Exome Research Panel v2.0 捕获探针(IDT,美国)与 gDNA 文库序列进行液体杂交,对目标区域 DNA 片段进行富集,构建全外显子文库;使用 DNBSEQ-T7 平台(华大智造,中国)进行测序,目标序列测序覆盖度不低于 99% 并对测序原始数据进行质控;使用 Burrows-Wheeler Aligner(BWA)软件将测序序列与 Ensemble 参考基因组 GRCh 37/hg 19 比对;然后使用 GATK 软件分析出单核苷酸多态性(SNP)、插入缺失变异;最后根据测序深度、突变质量对检测到的 SNP、插入缺失变异进行过滤和筛选,得到高质量的突变;使用变异注释软件(智因东方,中国)对检测到的高质量变异进行多种数据库(包括 dbSNP、千人基因组、ExAC、ESP、OMIM、HGMD、ClinVar 等)的关联注释。借助蛋白结构预测软件、剪切位点预测软件等对其危害性进行分析,筛选出对蛋白结构可能有有害影响的变异,并给出致病性评估。
根据变异位点设计相应的引物:正向引物 5'-CGCCCAGGCTTACATGTTTTATTT-3',反向引 5'-GAACTGCTCCCTACACAGGATAGA-3';采用 PCR 方法进行扩增;扩增的 PCR 产物经 1% 琼脂糖凝胶电泳验证,使用 ABI 3730 XL 测序仪(ABI,美国)进行 Sanger 测序,验证 PSAT1 基因变异位点并明确变异来源;采用 DNASTAR 软件进行序列分析和比对。
1.2.3 蛋白质三维结构预测 在 UniProt 数据库中下载 PSAT 的氨基酸序列,根据基因变异信息得到突变蛋白的氨基酸序列;使用 AlphaFold Server 软件分别预测蛋白质变异前后的结构,并使用 PyMOL v3.1.3 对置信度 pLDDT 最高的一组预测模型进行分析。
以「PSAT1」为关键词检索 UniProt 数据库,查找 PSAT 在不同物种中的氨基酸序列,并比较基因变异位置及前后的氨基酸序列差异情况。
1.2.4 文献复习 以「PSAT1」 or "Serine deficiency disorders"为关键词检索 PubMed 数据库;以「PSAT1」或「丝氨酸缺乏症」为关键词检索万方数据库和中国知网;设置发表时间年限为 20 年,末次检索时间为 2025 年 12 月。在与 PSAT1 基因变异有关的丝氨酸缺乏症文献中,选取相符的病例进行总结分析。
2 结果
2.1 临床资料
患儿,女,12 岁。出生时无明显皮肤异常,幼时走路易摔倒,2 岁时不明诱因下出现全身皮肤干燥脱屑,表现为躯干、四肢部位脱屑。7 岁时掌跖部位皮肤角化、发红、脱屑,并出现足部行走无力。症状逐渐加重,后发展为双下肢无力(下蹲后无法站起)。2025 年 7 月就诊于我院皮肤科,体格检查示神清,精神可,全身浅表未触及肿大淋巴结;神经系统检查示双侧轻度眼睑下垂及轻微水平眼震,双侧鼻唇沟对称,四肢肌张力正常,近端肌力 5 级,远端肌力 4 级,双侧小腿肌肉明显萎缩,膝反射亢进,踝反射正常引出,行走步态异常,表现为跑步慢及下蹲后无法自行站立;足部检查见双侧足跖不整齐伴明显高足弓;皮肤检查见掌跖部红斑角化,躯干及四肢部位皮肤干燥脱屑。
既往外院检查结果血常规、肝肾功能、尿便常规、心肌酶谱、血电解质、凝血功能等常规实验室检查均未见明显异常;脑电图显示α波率慢,稍多慢波,左额区稍著 ;神经超声显示双侧腓总神经、腓肠神经内结构欠清,腓总神经经腓骨小头处神经内回声稍减低,右侧腓肠神经横截面积较对侧稍大;双侧小腿 MR 平扫显示双侧小腿肌肉 MR 改变,以后群脂肪浸润及水肿改变为主。本院检查结果脑脊液、血清周围神经病变抗体阴性;肌电图显示左拇短展肌、右伸指总肌、股四头肌、双胫前肌呈神经源性损害;肌肉活检病理发现骨骼肌存在破碎红纤维和 SSV 现象,提示线粒体代谢异常可能。其间检测血浆丝氨酸水平正常:2022 年 233.29μmol/L、2025 年 319.19μmol/L(正常范围 86~500μmol/L)。
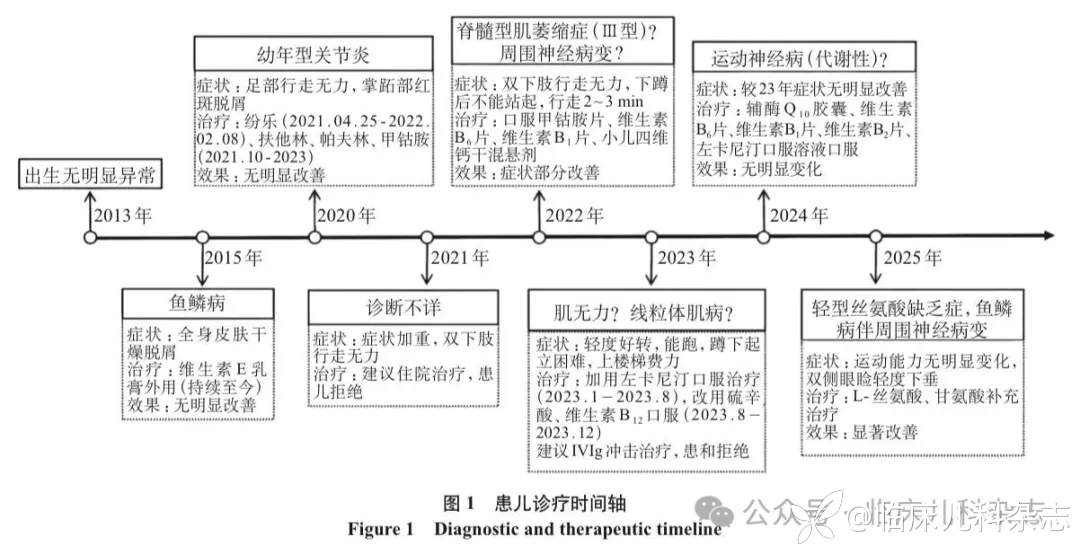
病程中除曾按「鱼鳞病」诊断,遂予维生素 E 乳膏外用治疗。随后因「足部行走无力,逐渐加重」,拟诊「幼年型关节炎」,先后尝试纷乐免疫抑制、扶他林和帕夫林抗炎、甲钴胺等治疗,均无明显效果。后续考虑「周围神经病变?」,给予营养神经和代谢辅助治疗后症状进展减缓。进一步根据肌肉活检结果考虑「线粒体肌病」,但进行家系全外显子组测序、线粒体基因组基因检测等多项遗传学检测,均未发现明确致病性变异。同时患儿双下肢无力症状仍持续进展,运动功能无明显改善,近一年维持可独立行走、上楼需扶、蹲下无法站起的状态,皮肤症状持续存在。患儿诊疗时间轴见图 1。
2.2 遗传学检测结果
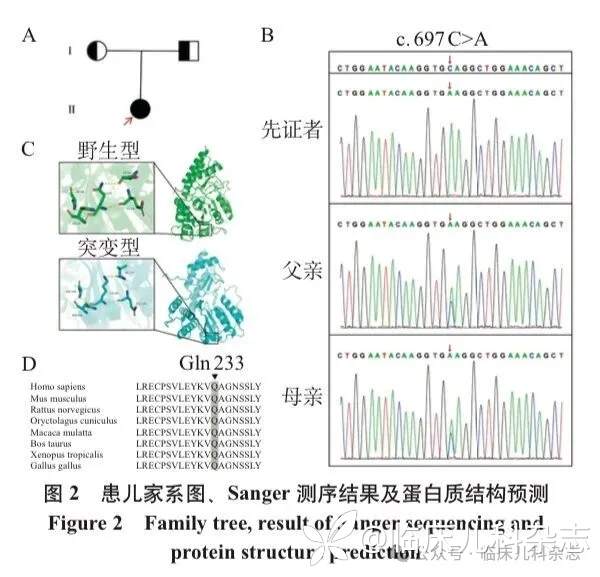
复查家系全外显子组测序数据,并使用 Sanger 测序进行验证,发现患儿携带 PSAT1 基因 c.697C>A(p.Q 233 K)纯合变异,ACMG 评级为意义不明确(VUS),患儿父母均为携带者(图 2A,B)。该变异在人类基因变异数据库(HGMD)、百万中国人群基因变异数据库(CMDB)和临床变异数据库(ClinVar)中均未记录。使用 AlphaFold Server 分别预测蛋白质变异前后的结构,分析显示变异导致第 233 位谷氨酰胺变为赖氨酸,谷氨酰胺原本的侧链酰胺氮原子与第 226 位丝氨酸的盐桥消失,取而代之的是赖氨酸伯胺侧链与第 227 位缬氨酸的作用力(图 2C)。保守性分析显示第 233 位的谷氨酰胺在不同物种中具有高度的保守性(图 2D)。结合临床表型,拟诊「轻型丝氨酸缺乏症,鱼鳞病伴周围神经病变」。
2.3 治疗及随访
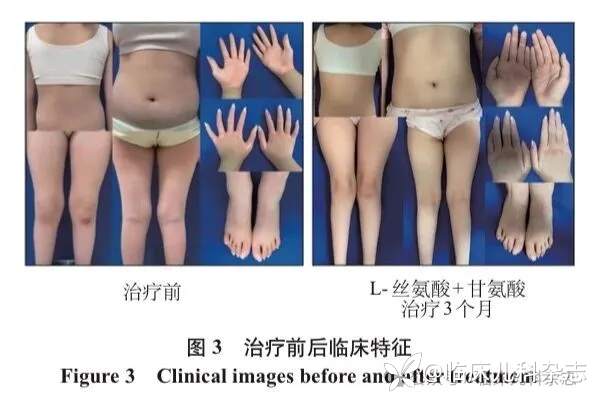
给予患者口服 L-丝氨酸(300 mg·kg-1·d-1)和甘氨酸(200 mg·kg-1·d-1)补充治疗,同时继续营养神经治疗:口服辅酶 Q10、维生素 B1 片、维生素 B2 片、左旋肉碱、维生素 C、维生素 E。每周随访 1 次,随访内容包括皮肤(角化、发红、脱屑)和周围神经病(行走步态异常和下蹲后蹲起障碍)改善情况。治疗 1 周后皮肤干燥脱屑明显好转,掌跖部鳞屑减少,红斑轻度改善 ;治疗 5 周后掌跖部红斑角化明显好转,行走步态轻度改善;治疗 13 周后行走步态明显改善,下蹲后可自主站起。后续仍需持续治疗并随访,监测皮肤、运动功能的改善情况及有无副作用(图 3)。
2.4 文献复习
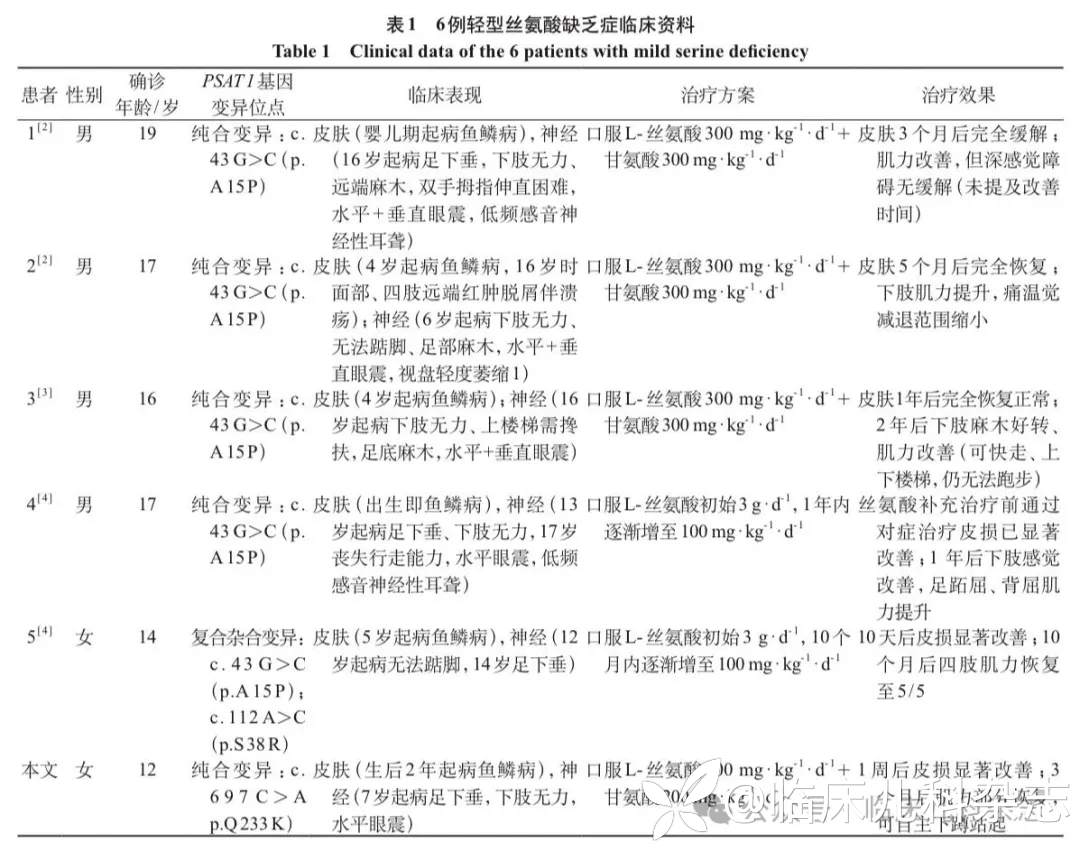
检索到相关中英文文献 3 篇,包含 5 例患者,所有患者均为 PSAT1 基因变异,且临床特征均表现为儿童期鱼鳞病和青少年期周围神经病,无中枢神经系统受累及生长受限。经口服 L-丝氨酸(单用或联合甘氨酸)治疗后,所有皮肤症状均显著改善,但神经症状改善程度因治疗年龄不同而存在差异,部分患者暂未恢复全部功能。本文新发现 1 种 PSAT1 基因错义变异(c.697C>A),汇总见表 1。
3 讨论
L-丝氨酸是一种非必需氨基酸,是多种必需化合物的前体,在胚胎发育、神经元功能、皮肤生理功能中具有重要作用 [5]。L-丝氨酸的主要来源是机体从头合成,也可通过饮食摄入、蛋白质降解或甘氨酸转化等途径获取。丝氨酸经典生物合成的第一步是 3-磷酸甘油酸在 PGDH 催化下转化为 3-磷酸羟基丙酮酸;第二步通过 PSAT 作用生成 3-磷酸丝氨酸;最后经 PSP 去除磷酸基团形成 L-丝氨酸 [1]。
丝氨酸缺乏症是一类由 L-丝氨酸生物合成通路中 3 种酶(PGDH、PSAT、PSP)缺陷导致的遗传性代谢病。任一关键酶的功能缺失,均可导致广泛的表型谱 [5]。根据临床表型的严重程度,我们将丝氨酸缺乏症分为致死型、非致死型和轻型。表型的严重程度主要取决于残存酶活性的高低:纯合或复合杂合截短突变(无义或移码)仅见于最严重的致死型,提示酶活性几乎完全丧失;而错义突变可见于各种表型,其严重程度与计算机预测的致病性评分相关;非致死型患者的成纤维细胞酶活性通常为正常水平的 10%~25%,而致死型的酶活性推测低于此范围 [6]。致死型丝氨酸缺乏症即 Neu-Laxova 综合征(Neu-Laxova syndrome,NLS)多在产前或出生后早期死亡,90% 以上为 PHGDH、PSAT1 基因变异致病,典型临床特征包括神经系统畸形、特征性面部畸形、皮肤异常和肢体异常 [7-9]。非致死型丝氨酸缺乏症可由 PHGDH、PSAT1、PSPH 基因变异致病,典型临床特征包括神经系统异常和生长相关症状,一般无多系统严重畸形,此类患者可存活至青少年或成年,但需长期管理 [5,10-12]。轻型丝氨酸缺乏症由 PSAT1 特定变异致病,典型临床特征包括皮肤异常(鱼鳞病)和神经系统表现(青少年起病周围神经病),无明显中枢神经系统症状及生长受限,该病症状轻微,极易漏诊,补充丝氨酸治疗后预后较好 [2-4]。此外,修饰基因和母体丝氨酸水平等环境因素也可能对表型产生一定影响 [5]。
对轻型丝氨酸缺乏症的诊疗要注意:首先,对于儿童期出现非典型鱼鳞病,尤其伴进行性下肢无力、眼震或低频感音神经性耳聋者,应警惕本病可能。其次,因膳食中摄入的氨基酸可使患者非空腹状态下的血浆丝氨酸及甘氨酸浓度表现为正常,因此即使血浆丝氨酸水平正常,亦不能排除该病诊断,需及时进行脑脊液氨基酸分析和基因检测以明确病因 [6]。最后,确诊后应尽早启动口服 L-丝氨酸补充(可联合甘氨酸以优化疗效)治疗,皮肤症状通常迅速缓解,而神经功能恢复则需长期坚持治疗。
轻型丝氨酸缺乏症目前仅有 6 例报道所有患者均为 PSAT1 基因变异致病,其中 4 例患者为纯合变异 c.43 G>C(p.A15P),1 例患者为复合杂合变异 c.43 G>C(p.A15P);c.112A>C(p.S38R),本文病例为纯合变异 c.697C>A(p.Q233K)。所有患者均表现出典型的儿童期鱼鳞病和青少年期周围神经病,确诊年龄集中在青少年时期。皮肤表现主要为全身干燥、脱屑,个别患者(1/6)有面部、四肢远端的皮肤溃烂,我们推测该表型可能与末梢感觉神经麻木相关。出现神经系统表现一般集中在 12~16 岁,本文病例较早,7 岁开始发病。所有患者均有进行性周围神经病变表现,如下肢无力、足下垂、远端感觉障碍等,严重者(1/6)丧失行走能力。多数患者(5/6)还伴有水平眼震,其中 3 例合并垂直眼震,2 例合并低频感音神经性耳聋,1 例合并视盘轻度萎缩。所有患者在单纯口服 L-丝氨酸(2/6)或与口服甘氨酸联合治疗(4/6)后,皮肤症状数月内均出现显著改善,而神经症状改善较为缓慢,持续治疗 1 到 2 年肌力部分恢复,且治疗启动年龄与神经功能预后密切相关:治疗越晚,神经功能恢复越差,严重者可能无法恢复全部功能。这一现象强烈提示早期诊断和干预对预防不可逆神经损伤具有决定性意义。
当前研究尚存在一定局限性。由于目前已报道的病例极为有限,难以开展系统性临床研究。且现有报道随访时间较短,对治疗后的远期预后认识尚不充分,基因型与表型之间的关联尚未完全阐明,尽管轻型丝氨酸中 PSAT1 基因不同变异所致临床表型呈现较高的一致性,但其背后的分子机制仍缺乏功能性研究的验证。尽管目前治疗方案有效,但最佳剂量、疗程及联合用药方案尚需更多临床数据支持。综上所述,丝氨酸缺乏症表型谱广泛,其中轻型丝氨酸缺乏症以儿童期鱼鳞病及青少年期周围神经病为特征,且血浆丝氨酸水平多正常或轻度降低,极易漏诊。提高临床识别能力、尽早启动 L-丝氨酸补充治疗是改善预后的关键,可避免不可逆神经损伤。
好文章,需要你的鼓励