



来源:临床儿科杂志
作者:黄文琛 1 冯艺杰 2 王肖艺 2 姚 妹 3 毛姗姗 2
作者单位:浙江大学医学院附属儿童医院 儿童少年健康与疾病国家临床医学研究中心 1. 超声科,2. 神经内科,3. 感染科
基金项目:国家自然科学基金项目
通信作者:毛姗姗 电子信箱:6307003@zju.edu.cn
推荐引用格式:
黄文琛, 冯艺杰, 王肖艺, 等. 外显子 1 结构变异致脊髓性肌萎缩症 2 例临床分析并文献复习 [J]. 临床儿科杂志, 2026, 44(3): 185-191 DOI:10.12372/jcp.2026.25e0645
HUANG Wenchen, FENG Yijie, WANG Xiaoyi, et al. Clinical analysis of two cases of spinal muscular atrophy caused by structural variations in exon 1 and literature review[J]. Journal of Clinical Pediatrics, 2026, 44(3): 185-191 DOI:10.12372/jcp.2026.25e0645
摘要
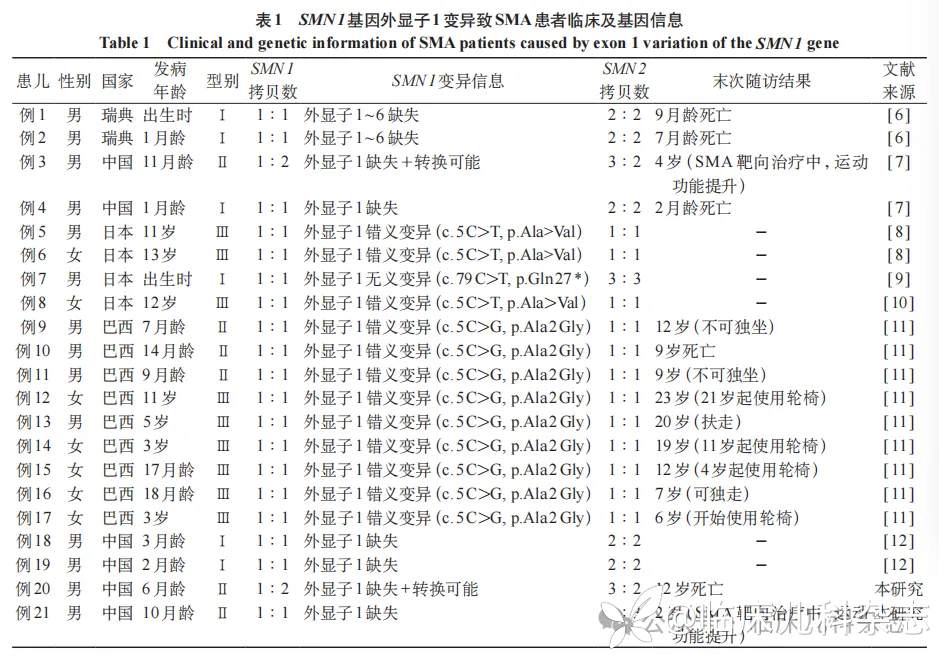
目的 总结 2 例运动神经元存活基因 1(SMN1)外显子 1 结构变异致脊髓性肌萎缩症(SMA)的临床表型和基因型特点。方法 回顾性分析 2022 年 10 月至 2024 年 6 月在神经内科就诊的 2 例外显子 1 结构变异致 SMA 患儿的临床资料,采集患儿及父母的外周血样,最终经长片段 PCR 技术结合多重连接探针扩增技术(MLPA)-P021 明确 5qSMA 诊断,并对 SMN1 基因外显子 1 变异致 SMA 相关文献进行总结分析。结果 2 例外显子 1 结构变异致 SMA 患儿均为男性,Ⅱ型,发病年龄分别为 7 月龄及 9 月龄,就诊年龄分别为 9 岁 1 月龄及 1 岁 1 月龄,均表现为四肢肌无力和运动发育落后,肌电图均提示脊髓前角病变。2 例患儿常规 MLPA 检测均为 SMN1 基因杂合缺失,最终经长片段 PCR 结合 MLPA-P021 确诊,检测结果均显示 SMN1 基因外显子 7 拷贝数为 1,同时该等位基因上检测到外显子 1 结构缺失变异。此外,例 1 患儿的该等位基因可能为 SMN1/SMN2 转换基因。例 1 患儿出生时间较早且其基因转换并结构变异较为罕见,诊断时间窗长达 11 年。文献检索共纳入 7 篇外显子 1 变异致 SMA 相关文献,包括错义变异、无义变异及结构缺失变异等,共 19 例,男女比 12∶7。患者在出生到青春期不同年龄起病,主要表现为运动功能落后或退步,其中 5 例已宣告死亡,部分接受 SMA 精准治疗的患者情况尚可。结论 该研究 2 例 SMA 患儿均为 SMN1 基因外显子 1 结构变异所致 5qSMA。当临床疑似 SMA 患者初次 MLPA 检测为阴性即外显子 7 未出现纯合缺失时,需考虑到 SMN1 基因外显子 1 变异,通过优化的长片段 PCR 结合 MLPA-P021 技术可快速精准明确诊断。外显子 1 可能为中国 SMA 变异图谱热点,同时基因转换值得关注。
关键词
脊髓性肌萎缩症; 复合杂合变异; 外显子 1; 长片段 PCR; 儿童
脊髓性肌萎缩症(spinal muscular atrophy,SMA)是一种常染色体隐性遗传性疾病,主要表现为进行性肌无力和/或肌萎缩,常累及呼吸、消化、骨骼等多个系统,位居 2 岁以内致死性遗传病首位 [1]。SMA 致病基因为运动神经元存活基因 1(survival motor neuron gene 1,SMN1)[2],发病率约为 1/10000,携带率约为 1/50[3]。依据国际 SMA 多学科诊治共识推荐 [4],目前 SMA 基因诊断主要通过多重连接探针扩增技术(multiplex ligation-dependent probe amplification,MLPA)或定量聚合酶链式反应技术(quantitative polymerase chain reaction,qPCR)来明确 SMN 基因拷贝数,该检测方法可确诊约 95% 的 SMN1 纯合缺失患者。当 SMN1 拷贝数为 1 或 2 时,需再通过长片段 PCR-巢式 PCR 或逆转录-克隆测序以明确 SMN1 基因是否存在其他变异 [3]。本研究报道了 2 例经初次 MLPA 检测未能明确的 SMN1 基因外显子 1 结构变异致 SMA 患儿,同时检索相关文献进行总结,以期为罕见 SMA 的精准诊断提供更多依据。
1 对象与方法
1.1 研究对象
回顾性分析 2022 年 10 月至 2024 年 6 月在浙江大学医学院附属儿童医院神经内科诊断的 2 例外显子 1 结构变异致 SMA 患儿的临床资料及遗传学检测结果。
1.2 方法
1.2.1 临床资料收集 包括患儿的基本信息(性别、起病年龄、生长发育史、家族史等),体格检查,诊断治疗经过,预后等临床信息,以及实验室检查、肌电图检查、基因检测等检查结果。
1.2.2 文献检索 计算机检索 PubMed、Web of Science、Cochrane Database、Embase、CNKI、WanFang Data 和 CBM 数据库,搜集有关 SMN1 基因外显子 1 变异致 SMA 的文献报道,设置发表时间年限为 20 年,末次检索时间为 2025 年 5 月。检索采取主题词和自由词结合的方式。英文数据库检索以「spinal muscular atrophy」「composite heterozygous variation」「exon 1」「SMN gene conversion」为主题词进行检索;中文数据库检索用「脊髓性肌萎缩症」「复合杂合变异」「外显子 1」「SMN 基因转换 」进行主题词和自由词检索。
2 结果
2.1 临床资料
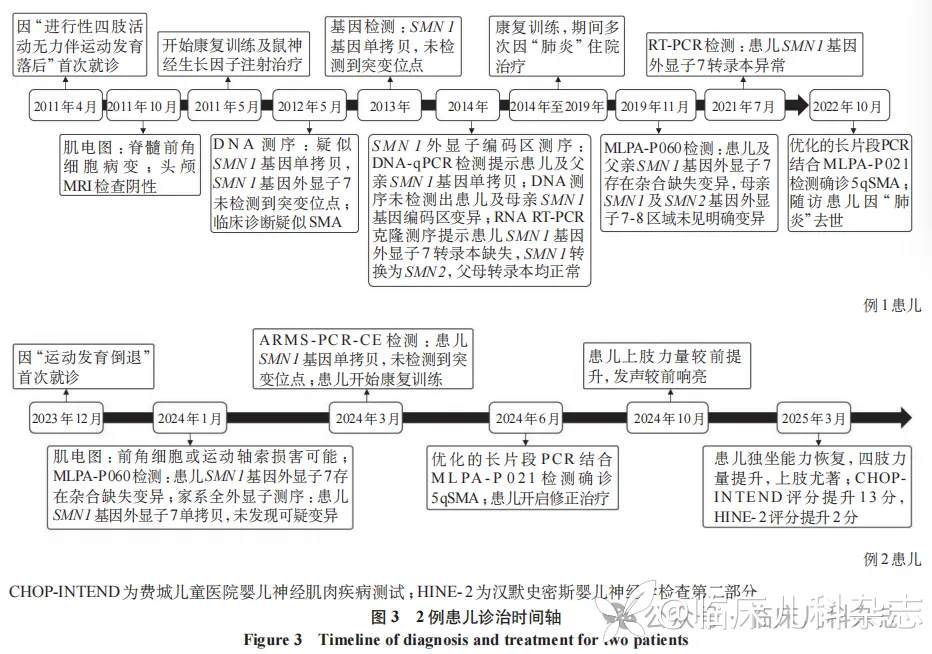
例 1,男,9 岁 1 月龄,因「进行性四肢肌无力伴运动发育落后 8 年余」于 2019 年 11 月就诊于浙江大学医学院附属儿童医院神经内科。7 月龄时家长发现患儿双下肢活动无力,运动发育落后于同龄儿,无法完成翻身及爬行动作。18 月龄时患儿可独坐,至今不能独走。患儿系 G1P1,足月自然分娩,否认出生窒息抢救史。父母体健,否认近亲婚配史,弟弟(5 岁)无类似症状,否认家族遗传病史。体格检查:神清,精神可,颈软,双上肢肌力Ⅲ级,双下肢肌力Ⅱ级,四肢肌张力低下,双侧跟膝腱反射未引出,双侧巴氏征阴性。辅助检查:血常规、生化、血气和电解质等结果均正常,肌电图提示脊髓前角细胞病变。患儿先后至全国多家医院就诊,先后检测 DNA 测序提示疑似 SMN1 基因单拷贝,SMN1 外显子编码区测序、RNA 反转录 PCR(RT-PCR)克隆测序及反转录定量 PCR(RT-qPCR)提示转录本异常,初次 MLPA 检测提示 SMN1 基因外显子 7 杂合缺失,全外显子测序(WES)结果阴性,全基因组测序结果阴性,均未能获得明确诊断。
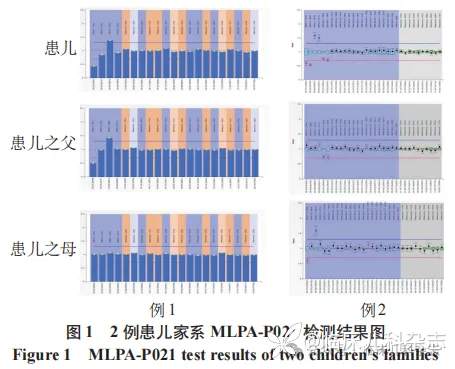
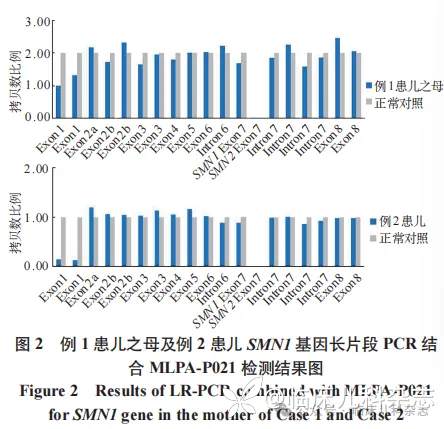
MLPA-P021 检测结果(图 1)显示患儿 SMN1 基因存在杂合缺失变异,SMN1 基因外显子 7 和 8 的拷贝数分别为 1 和 2,SMN2 基因外显子 7 和 8 的拷贝数分别为 3 和 2,可能存在 SMN 基因转换,SMN1 及 SMN2 基因外显子 1 拷贝数总和为 3,根据外显子 7、8 拷贝数一致且总和为 4,该患儿 SMN 基因的外显子 1 存在 1 个拷贝的缺失。SMN2 基因长片段 PCR 结合 MLPA-P 021 检测结果显示患儿在 SMN2 基因上无外显子 1 拷贝数结构缺失。患儿父亲的 SMN1 基因外显子 7 和 8 的拷贝数分别为 1 和 2,SMN2 基因外显子 7 和 8 的拷贝数分别为 3 和 2,可能存在基因转换。患儿母亲的 SMN1、SMN2 基因外显子 7、8 拷贝数均为 2,SMN1 及 SMN2 基因外显子 7、8 总拷贝数为 4,外显子 1 拷贝数总和为 3,存在 1 拷贝的缺失。SMN1 基因长片段 PCR 结合 MLPA-P021 检测结果提示患儿母亲外显子 1 结构缺失发生在 SMN1 基因上(图 2)。
例 2,男,1 岁 1 月龄,因「运动发育倒退 4 月余」 于 2024 年 6 月就诊于浙江大学医学院附属儿童医院神经内科。患儿出生后早期运动发育正常,9 月龄时出现运动能力倒退,表现为抬头困难,独坐不稳,双下肢显著无力,无法完成踢腿动作,病初至外院康复治疗,就诊时患儿可抬头,可独坐 10 分钟,无法完成翻身、爬行动作。患儿系 G1P1,足月自然分娩,否认出生窒息抢救史。父母体健,否认近亲婚配史,否认家族遗传病史。体格检查 :神清,精神可,颈软,双上肢肌力Ⅲ级,双下肢肌力Ⅱ级,四肢肌张力低下,双侧跟膝腱反射未引出,双侧巴氏征阴性。辅助检查:血常规、生化、血气和电解质等结果均正常,肌电图提示神经源性损害累及上下肢,前角细胞或运动轴索损害可能。外院 MLPA-P060 检测结果提示 SMN1 基因杂合缺失,WES 提示 SMN1 单拷贝,未见点突变。
扩增阻滞突变系统聚合酶链反应-毛细管电泳(amplification refractory mutation system polymerase chain reaction-capillary electrophoresis,ARMS-PCR-CE)检测 [5] 显示患儿 SMN1 基因外显子 7、8 拷贝数均为 1,SMN2 基因外显子 7、8 拷贝数均为 3,且未发现点突变。MLPA-P021 检测结果(图 1)显示患儿 SMN1/2 基因外显子 7、8 拷贝数同前,SMN1 及 SMN2 基因外显子 7 和 8 总拷贝数为 4,外显子 1 总拷贝数为 3,根据总拷贝数一致性,SMN 基因外显子 1 存在 1 个拷贝数的缺失变异。患儿父亲的 SMN1 基因外显子 7、8 拷贝数均为 2,SMN2 基因外显子 7、8 拷贝数均为 2,SMN1 及 SMN2 基因外显子 1 总拷贝数为 3。患儿母亲的 SMN1 基因外显子 7 拷贝数为 1、外显子 8 拷贝数为 2,SMN2 基因外显子 7 拷贝数为 3、外显子 8 拷贝数为 2,可能存在基因转换。患儿长片段 PCR 结合 MLPA-P021 检测结果(图 2)显示 SMN1 基因存在外显子 1 结构缺失。
2 例患儿的诊治时间轴见图 3。依据美国医学遗传学与基因组学学会标准进行评估,上述变异均被判定为致病性变异,2 例患儿均确诊为Ⅱ型 SMA 患儿。
2.2 文献检索结果
检索 SMN1 基因外显子 1 变异致 SMA 的相关文献,查询到 7 篇 [6-12] 共报道 19 例患者,加上本研究 2 例,共计 21 例,男 14 例、女 7 例。患者在出生到青春期不同年龄起病,主要表现为运动功能落后或退步,临床类型包括Ⅰ~Ⅲ型,其中我国报道 6 例,均涉及外显子 1 结构缺失。临床及基因变异信息见表 1。5 例在研究中已报告死亡 [6-7,11],其中 3 例为Ⅰ型 SMA 患儿,2 例为Ⅱ型 SMA 患儿。我国报道的 2 例Ⅱ型患儿在确诊后进行精准治疗,运动功能均得到提升 [7]。
3 讨论
SMA 是一种罕见的常染色体隐性遗传性疾病,其主要特征为神经元细胞退化变性导致的进行性近端肢体和躯干肌无力和/或肌萎缩。该病的致病基因是位于染色体 5q13.2 区域的 SMN1[2]。95% 的 SMA 患者为 SMN1 基因纯合缺失,约 5% 患者为复合杂合变异类型 [3]。国际 SMA 多学科诊治共识 [4] 明确指出,临床疑似 SMA 的患者应首先进行 MLPA 检测。如果检测结果显示存在 SMN1 基因杂合缺失,则应进一步使用长片段 PCR 结合 Sanger 测序进行检测,以确定 SMN1 基因上是否存在其他变异。但在人体中同时存在与 SMN1 基因高度同源的 SMN2 修饰基因,只有 16 处碱基的差异 [13],这给部分 SMA 患者的诊断带来了较大困难,经常导致误诊、漏诊和延误治疗 [14-15]。
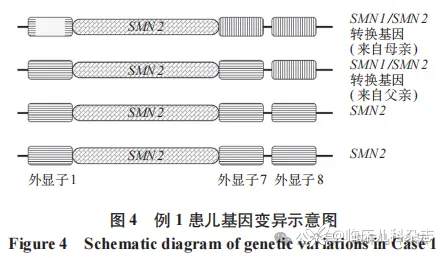
本研究中的 2 例患儿均表现为运动功能发育迟缓及退化,肌电图检查结果均提示存在神经源性损伤,临床上高度怀疑为 SMA。依据 SMA 诊断标准,2 例患儿及其父母均进行了 MLPA 检测,结果显示 2 例患儿的 SMN1 基因拷贝数均为 1,两例患儿可能为 [0+1d] 型 SMA,也可能仅为 SMA 携带者,未能明确诊断。参考 Qu 等 [16] 研究,检测例 1 患儿的 SMN1 基因 mRNA 转录本水平提示异常降低,进一步支持了 5qSMA 诊断。结合前期本团队诊断的 2 例 SMN1 基因外显子 1 缺失致 SMA 病例 [12],本研究同样关注了 2 例患儿 SMN1 基因外显子 1 的拷贝数。应用前期优化的长片段 PCR 结合 MLPA-P021 检测方法 [12],再次进行实验及分析显示,例 1 患儿的 SMN1 及 SMN2 基因中外显子 1 的总拷贝数为 3,提示外显子 1 存在缺失。该患儿母亲的外显子 1 拷贝数也存在缺失,且该缺失最终定位于 SMN1 基因。由于该患儿已死亡,遂根据家系遗传传递分析推测,该患儿可能发生基因转换导致的 SMN1 等位基因缺失遗传自其父亲,另一 SMN1 等位基因的外显子 1 缺失遗传自其母亲。此外,结合一代测序结果,例 1 患儿的该 SMN1 等位基因上可能存在外显子 1 至内含子 6 向 SMN2 基因转换的情况,推测该等位基因为 SMN1/SMN2 转换基因,其可能的基因型见图 4。同样地,通过该优化的基因检测方法,例 2 患儿家系的长片段 PCR 结合 MLPA-P021 检测结果提示其 SMN1 基因外显子 1 存在结构变异。根据家系遗传规律分析,该外显子 1 结构缺失变异遗传自患儿父亲,其母亲为 SMN1 基因外显子 7 杂合缺失类型的携带者。
在我国的 SMA 基因变异图谱中,外显子 1 的变异被认为是可能的热点 [17]。本研究的 2 例患儿为 SMN1 基因外显子 1 结构变异,同时例 1 患儿在该等位基因上可能还存在转换变异,结合临床表现均被确诊为Ⅱ型 SMA。当常规基因检测未能明确诊断,但临床表现符合 SMA 时,应考虑 SMN1 基因非外显子 7/8 变异或转换,尤其是热点变异外显子 1 异常 [7,12]。由于 SMA 完全由致病基因 SMN1 基因的变异引起,而 MLPA 无法区分外显子 1 具体位置,因此在检测时需要分离 SMN1 基因进行单独检测,以明确诊断。长读长测序作为更全面和精准的测序方法已在 SMA 的早期诊断中应用 [17],但测序费用较高,尚不能普及,2 例患儿未能完成三代测序验证。本团队优化后的长片段 PCR 结合 MLPA-P021 的诊断方法利用长片段 PCR 特异性扩增 SMN1 基因,再结合 MLPA-P021 检测 SMN 基因外显子拷贝数,可一次性识别 SMN1 基因外显子 1 结构变异,相较于现有的 MLPA、qPCR 及 WES 等技术在速度、灵敏度、可靠性和可行性方面具有优势,同时获得了更好的经济效益,能够让更多的患儿家庭接受。通过我们优化的基因检测流程,2 例患儿已获得明确诊断,例 2 患儿实现了早诊早治、提高生活质量的目标。此外,例 1 患儿的母亲及例 2 患儿的父亲为隐匿性携带者 [1+1d],在通常的 SMA 携带者筛查中不能被发现,在后期的遗传生育咨询中需关注。
通过既往文献分析,SMA 的表型轻重与 SMN2 基因拷贝数的多少有关,SMN2 基因拷贝数越多,往往患者的临床表型越轻 [18]。然而,在这些外显子 1 变异致 SMA 的患者中,SMN2 基因拷贝数对表型的影响较小,似乎外显子 1 的变异比 SMN2 基因拷贝数对临床严重程度的影响更大,这一发现也与之前文献报道相符 [9]。本研究推测,与 SMN1 基因纯合缺失的患儿相比,具有外显子 1 缺失的复合杂合变异的患儿可能表现出相对较轻的症状,但仍需要通过更大的样本量和进一步的研究来验证这一假设。
目前,全球已上市三种 SMA 靶向治疗药物,其中反义寡核苷酸药物诺西那生钠(nusinersen)和小分子药物利司扑兰(risdiplam)也在国内上市并纳入医保,中国 SMA 患者治疗可及性得到极大提高。研究显示越早治疗,疗效越好,尤其是症状前的治疗可使患者获得最佳疗效,甚至达到与正常儿童类似的运动功能发育 [19],因此早期诊断尤为重要。例 1 患儿自 2011 年辗转全国多家医院未能明确诊断,主要进行康复训练,最终确诊时间窗约为 11 年,患儿在未经 SMA 精准治疗的情况下的存活时间为 12 年,最终因肺炎去世。得益于近几年诊断技术的快速发展,例 2 患儿确诊时间窗缩短为 6 个月,并在明确诊断后立即开始利司扑兰小分子口服药物治疗及康复训练,末次随访时已治疗 9 个月余,患儿的上肢力量提升显著,发声也较前响亮,运动功能得到显著改善,费城儿童医院婴儿神经肌肉疾病测试(children's hospital of Philadelphia infant test of neuromuscular disorders,CHOP-INTEND)评分从 44 分提升至 57 分,汉默史密斯婴儿神经学检查第二部分(Hammersmith infant neurological examination section 2,HINE- 2)评分从 10 分提升至 12 分。
自 2006 年 Arkblad 等 [6] 首次报道 SMN1 基因外显子 1~6 缺失变异,现共有 21 例涉及外显子 1 变异的 SMA 病例报道,因此,外显子 7/8 以外的其他外显子变异同样值得重视和关注。这些研究报道扩大了 SMA 变异图谱,使得 SMA 基因检测的范围也不再局限于外显子 7/8。同时,本研究通过分析 2 例 SMN1 基因外显子 1 结构变异致 SMA 患儿的临床表现及基因检测结果,支持外显子 1 变异可能为中国人群热点变异 [17]。当临床疑似 SMA 患儿的常规基因检测结果为阴性时,需考虑外显子 1 是否存在致病性变异。应用优化的长片段 PCR 结合 MLPA-P021 技术,可为无法通过常规检测诊断的临床疑似 SMA 患者的精准诊断提供可靠方法。
好文章,需要你的鼓励