



2026 年 3 月 14 日,深圳市妇幼保健院儿科顺利完成一例 20 月龄戈谢病(IIIc 型)患儿首剂酶替代治疗,这是本院针对该类罕见病患儿开展酶替代治疗的新尝试。
戈谢病(Gaucher disease,GD)是因β-葡萄糖脑苷脂酶(GBA)基因缺陷导致的常染色体隐性遗传性罕见病。GBA 基因缺陷会造成该酶活性下降,体内的细胞垃圾「葡萄糖脑苷脂」无法被分解,进而堆积在肝、脾、骨骼、肺、神经系统巨噬细胞的溶酶体内形成「戈谢细胞」,最终引发肝脾肿大、血小板减少等临床表现。
小可(化名)出生时便出现血小板减少,3 个月大时开始出现腹胀症状,起初家人以为是「积食」「胃肠功能紊乱」。直到 1 岁半体检时,医生发现其腹部膨隆如蛙腹,检查后显示肝脾重度肿大,经酶学和基因检测发现,患儿葡萄糖脑苷脂酶活性为 0.89umol/l/h(正常参考值:1.19-22.23umol/l/h),且 GBA 基因存在纯合变异,最终被确诊为戈谢病 IIIc 型。
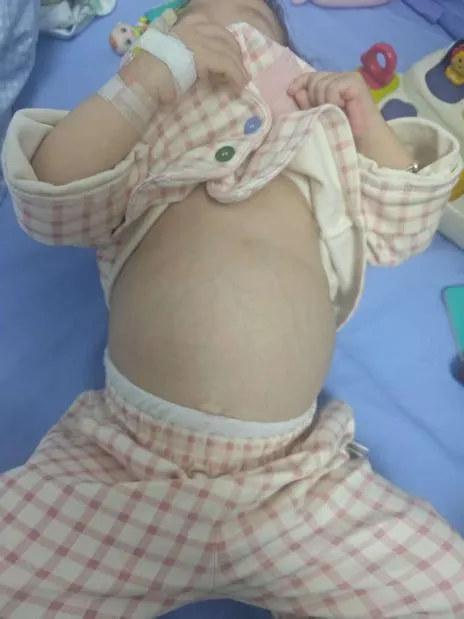
目前戈谢病尚无根治方法,幸运的是,定期接受酶替代治疗(Enzyme Replacement Therapy,ERT),能够帮助清除体内多余的葡萄糖脑苷脂,弥补体内该酶的缺陷并替代其发挥生理功能,助力细胞清除堆积的脂质,但该疾病需要终身用药。
为尽早实施酶替代治疗,阻止疾病进一步进展,医院第一时间安排小可住院完善相关检查,并对首次酶替代治疗的潜在风险及预后进行了充分评估,最终顺利为她完成首剂伊米苷酶替代治疗,治疗过程顺利,无任何不良反应。
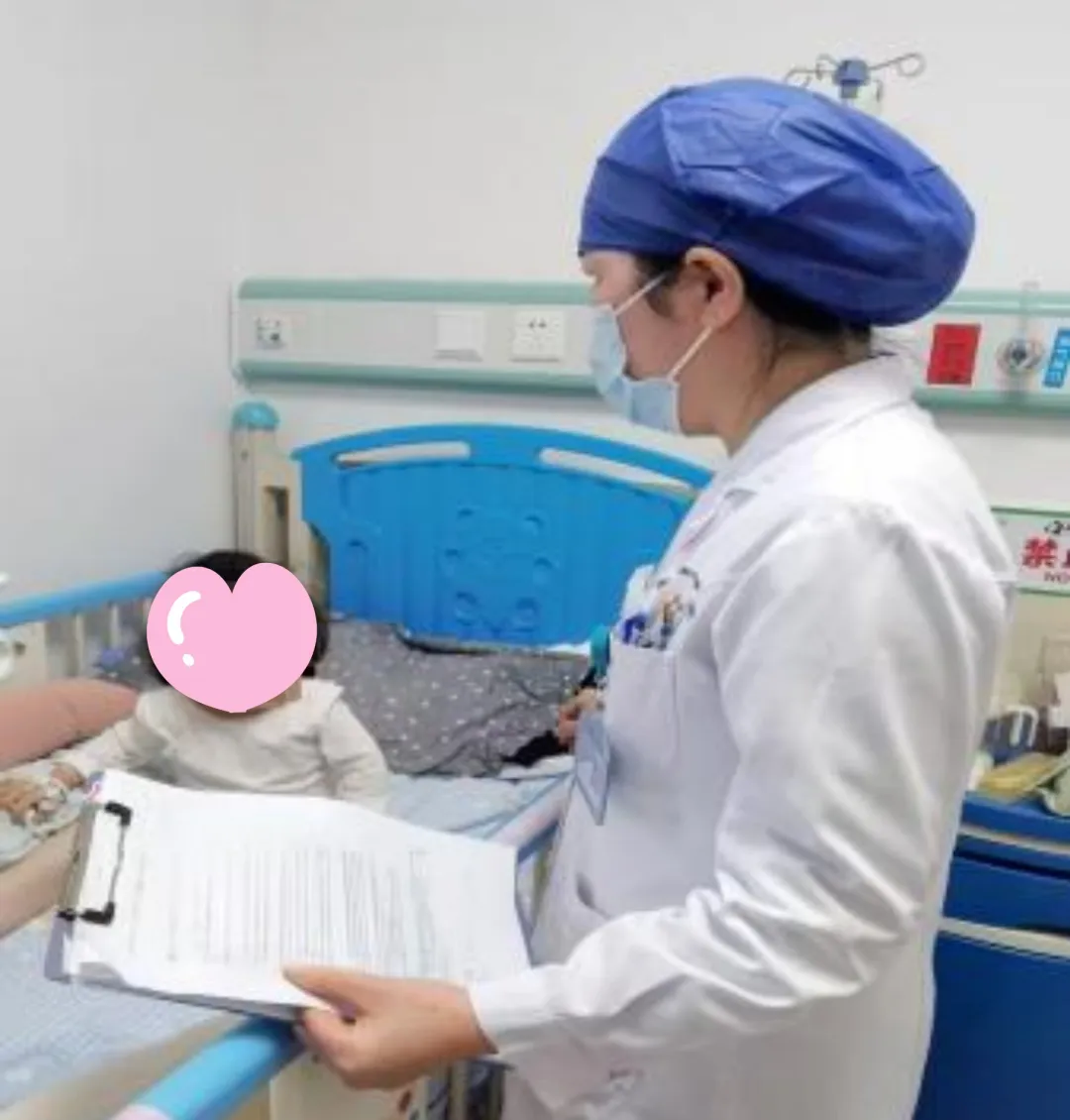
深圳市妇幼保健院针对罕见病还开设了遗传罕见病、神经罕见病、罕见病消化营养干预及再生育咨询等特色专科门诊,配齐专项检测与先进诊疗技术,联合生殖遗传 MDT 门诊实现罕见病的源头防控与代际阻断。
此外,科室持续联动国内顶尖专家罗小平教授团队开展学术交流,深耕罕见病临床研究与诊疗技术创新,以规范化的诊疗路径、全方位的医疗服务,为每一位罕见病患儿提供专业救治,用精准医疗为罕见病家庭点亮希望,让罕见病不再 「难诊难治」。
好文章,需要你的鼓励