



「医生,我连楼梯都爬不动了……」45 岁的张先生(化名)第一次走进神经内一科诊室时,呼吸费力、步履蹒跚。五年前,他因双腿酸胀、行走缓慢辗转多家医院,曾被诊断为「腰椎间盘突出」,但治疗毫无效果。随着时间的推移,张先生四肢无力的情况逐渐加重:无法跑步、提不起重物,甚至走几步就要休息。更揪心的是,他频繁地感到面部和双手麻木,近来更是出现夜间胸闷气短的症状,甚至难以呼吸。
在外院多次检查后,仅发现血钾低(3.2 mmol/L),但补钾治疗收效甚微。当张先生深受四肢无力病情困扰时,陕西省人民医院神经内一科王莉副主任医师团队通过抽丝剥茧,逐渐揭开了患者「四肢无力」背后的罕见病真相……
初诊迷雾——当「腰椎病」无法解释「进行性肢体无力」
5 年前,张先生因双下肢无力、行走缓慢就诊于当地医院。他描述自己「双腿像灌了铅」,但无麻木或疼痛。经腰椎 MRI 检查,结果显示「腰 4-5 椎间盘轻度膨出,腰椎骨质增生」。结合影像学报告,医生初步诊断为「腰椎退行性病变」,并给予药物保守治疗。然而,治疗数月后,张先生的症状不仅未缓解,反而逐渐加重。
腰椎间盘膨出,不足以解释患者四肢无力的症状。神经内一科王莉副主任医师分析道,「腰椎问题引起的神经压迫多表现为单下肢放射性疼痛伴行走跛行,而张先生的症状却是进行性四肢无力,且无典型神经根受压症状。所以,我们考虑该患者存在尚未被诊断的更深层的神经病理机制。」
为寻找答案,王莉团队展开首轮鉴别诊断。患者新斯的明试验和重频神经电刺激检查结果均为阴性,排除了重症肌无力;虽有低钾血症,但补钾后症状无改善,且无家族史,不符合低钾性周期性瘫痪;肌酶轻度升高,肌电图未见肌源性损害,因此多发性肌炎也被排除。
至此,常见肢体无力病因多被排除,王莉团队把诊断范围缩小至神经受累疾病。
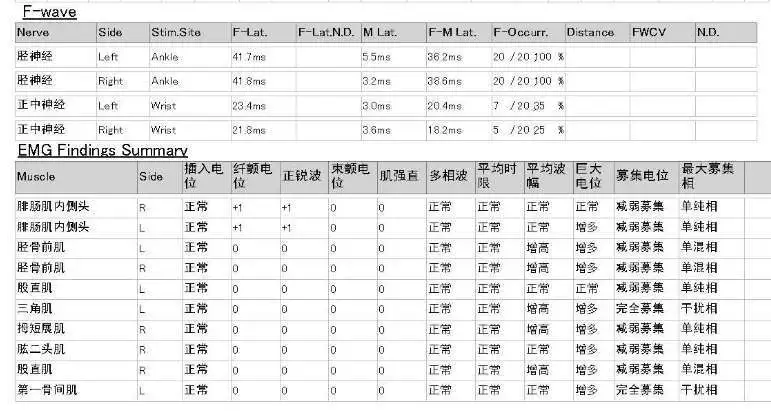
而在此时,患者再次复查的四肢肌电图为疾病的诊断带来了曙光。患者复查神经电生理检查结果显示颈髓、胸髓和腰髓支配肌肉同时可见活动期和慢性期神经源性损害。「我们当时考虑这可能是一例疑难罕见病。」王莉提到。
柳暗花明——基因检测破解「罕见病密码」
王莉解释,「肌萎缩侧索硬化症(Amyotrophic Lateral Sclerosis, ALS)、肯尼迪病(Spinal and Bulbar Muscular Atrophy, SBMA)及成人型脊髓性肌肉萎缩症(adult Spinal Muscular Atrophy,SMA-III 型)均可表现为逐渐进展的四肢无力,但三者的预后和治疗策略截然不同,需进一步鉴别。」
ALS、肯尼迪病还是 SMA-III 型?
考虑 ALS 多快速进展,平均生存期 3-5 年,而张先生病程长达 5 年,舌肌萎缩伴四肢近端无力进展缓慢,且伴有内分泌异常,因此王莉团队最终还是决定通过特定基因检测进一步明确诊断。张先生的雄激素受体(Androgen Receptor, AR)基因动态突变检测结果显示 CAG 三核苷酸重复次数为 45 次,超过正常阈值(正常 ≤ 34 次),因此张先生最终确诊为肯尼迪病。

据王莉介绍,肯尼迪病是一种罕见的、可伴全身多系统损害的、伴 X 染色体隐性遗传的神经肌肉疾病。我国发病率约为 2/100000,该病于 2018 年被纳入我国第一批罕见病名录。根据张先生缓慢进展的四肢近端无力、内分泌异常的临床表现,以及慢性神经源性损害的神经电生理特点和家族史,并结合相关检查结果,张先生的病情基本确诊。
多学科协作——从确诊到治疗的全程管理
根据指南和治疗方案,肯尼迪病目前尚无根治性疗法,主要目标是对症治疗及改善生活质量,包括肌力训练与康复、营养与吞咽指导;对于合并内分泌异常者可用抗雌激素治疗;支持性呼吸管理(严重脑干受累时)以及心理支持。

明确诊断后,王莉团队为张先生量身定制治疗方案。首先,指导张先生补充 B 族维生素,以进行营养神经治疗;其次,建议其加用亮丙瑞林,减少体内异常致病蛋白沉积,延缓肢体无力进展。
「康复与呼吸支持是本病治疗的重点。」王莉强调。针对张先生四肢无力和呼吸费力的问题,治疗团队联合康复科、呼吸与危重症科制定了个体化的长期康复方案,包括肌肉拉伸和低强度抗阻训练,以防止关节挛缩;指导腹式呼吸训练,储备呼吸功能;并要求张先生监测夜间低氧风险,必要时夜间佩戴家用呼吸机。

「肯尼迪病虽无法治愈,但病情进展缓慢,经过系统性治疗可以提高生活质量。」王莉为张先生讲解了疾病的特点及预后,帮他树立了强有力的信心。「感谢王莉医生团队的帮助,我知晓了自己患病的真相,现在反而没有那么恐慌、担心了。现在我的症状改善了很多,会积极配合治疗。」张先生感慨道。
王莉提及该罕见病的治疗过程时说道:「在临床工作的漫漫长路中,要关注患者的健康需求,从患者的角度出发,敢于迎接罕见病诊疗的挑战。努力扩充罕见病诊疗知识,不仅体现对患者生命的敬畏,更是医者不忘初心的追求。」
医疗科普
肯尼迪病科普
01 什么是肯尼迪病?
肯尼迪病是一种罕见的可伴全身多系统损害的伴 X 染色体隐性遗传的神经肌肉疾病,我国发病率约为 2/100000,于 2018 年被纳入我国第一批罕见病名录。
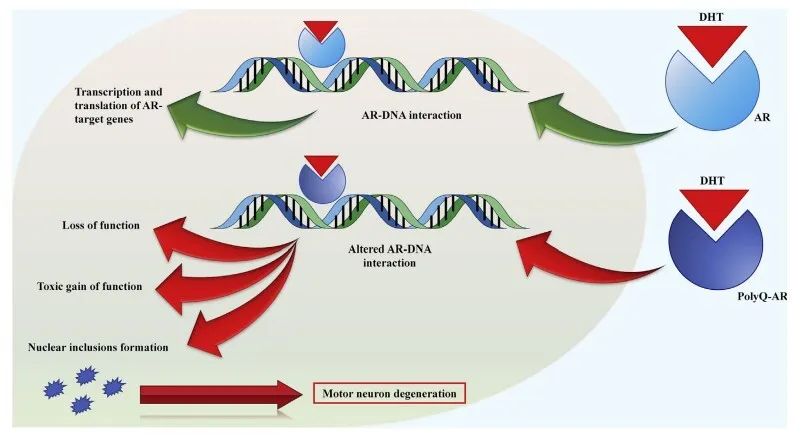
肯尼迪病由位于 X 染色体上的 AR 基因的 CAG 三核苷酸重复序列异常扩增引起。扩增的 CAG 序列编码多聚谷氨酰胺,形成构象异常的 AR 蛋白,这些蛋白在雄激素作用下积聚于细胞核内,形成毒性聚集体,导致脊髓前角和脑干的α运动神经元变性、肌肉萎缩与去神经化、周围神经病变、雄激素信号通路受损。
02 肯尼迪病有哪些症状?
患者多于 30–60 岁之间发病,起病隐匿,进展缓慢,仅男性发病,女性携带者多无症状或症状轻微。
神经肌肉表现为四肢近端肌无力与萎缩,以下肢为著,伴肢体麻木及神经性疼痛,伴舌肌萎缩、舌肌颤动、构音困难、吞咽困难、双手震颤、面肌无力、颌面部抽动;患者可出现乳腺发育、性欲减退、不育等内分泌表现;可能合并如汗液减少、便秘等自主神经功能障碍。
03 如何确诊肯尼迪病?
根据缓慢进展的四肢近端无力、内分泌异常的临床表现、慢性神经源性损害的神经电生理特点及家族史,可疑诊肯尼迪病。患者确诊需进行 AR 基因三核苷酸 CAG 重复次数检测,重复次数 ≥ 38 可确诊。
好文章,需要你的鼓励