



来源:临床儿科杂志
作者:伋自晶 1 黄海生 2 赵安琪 2 何 伟 2 李 敏 1 李 明 2
作者单位:1. 苏州大学附属第四医院(苏州市独墅湖医院)皮肤科;2. 复旦大学附属儿科医院(国家儿童医学中心)皮肤科
通信作者:李明 电子信箱:mingli@fudan.edu.cn
推荐引用格式:
伋自晶, 黄海生, 赵安琪, 等. 切-希二氏综合征 1 例报告 [J]. 临床儿科杂志, 2026, 44(5): 453-455 DOI:10.12372/jcp. 2026.25e1602
JI Zijing, HUANG Haisheng, ZHAO Anqi, et al.A case report of Chediak-Higashi syndrome[J].Journal of Clinical Pediatrics, 2026, 44(5): 453-455 DOI:10.12372/jcp.2026. 25e1602
摘要
目的 探讨切-希二氏综合征(Chediak-Higashi 综合征)的临床特点和遗传学特征。方法 采用家系全外显子组测序(trio-WES)鉴定致病基因,总结患儿临床资料,分析临床及遗传学特征。结果 患儿,男,1 岁 7 个月,以「面部、四肢远端色素沉着,伴灰发 1 年余」为主诉就诊于复旦大学附属儿童医院皮肤科。Trio-WES 检测发现 LYST 基因存在复合杂合变异,为 c.2962C>T,c.10564+1dupG,其中剪接位点变异为新发 (de novo) 变异,符合常染色体隐性遗传模式,最终诊断为 LYST 基因变异导致的切-希二氏综合征。结论 LYST 基因变异案例目前报道较少,本案例鉴定的变异是未见报道的,扩充了 LYST 基因缺陷的基因型-表型谱,也为进一步了解切-希二氏综合征提供数据。精确诊断依赖分子遗传学检测,需积累更多病例进一步分析基因型-表型关系和预后评估。
关键词
LYST 基因; 切-希二氏综合征; 全外显子组测序
引言
切-希二氏综合征(Chediak-Higashi 综合征,CHS)是一种罕见的常染色体隐性疾病,该病在临床表现上呈谱系分布,主要特征包括色素减退、反复细菌感染、发育迟缓及凝血功能异常 [1-3]。超过 80% 的患者会进入疾病加速期,临床表现为危及生命的噬血细胞性淋巴组织细胞增多症(HLH)——一种由病毒、细菌、真菌、寄生虫感染,或恶性肿瘤等多种原因诱发的过度炎症综合征。此加速期现象是 CHS 患者最常见的致死原因。CHS 的一个关键诊断特征是外周血涂片中白细胞内颗粒增大。另外也可能存在中性粒细胞减少以及自然杀伤细胞(NK)数量和功能减少 [4]。
CHS 的遗传学基础已被追溯至 LYST(溶酶体运输调节因子)基因的变异,该基因亦被称为 CHS1。LYST 基因位于染色体 1q42-43 区域,其编码的蛋白质参与调控溶酶体的大小、数量及功能。该基因发生变异会破坏正常的溶酶体运输过程,导致形成体积异常增大且功能失调的溶酶体。这些异常的溶酶体损害了多种关键的细胞过程,包括免疫应答、色素沉着及神经功能。迄今为止,已在 LYST 基因中发现了超过 60 种不同类型的变异,且大多数患者表现为新发变异或复合杂合变异 [5]。
1 临床资料
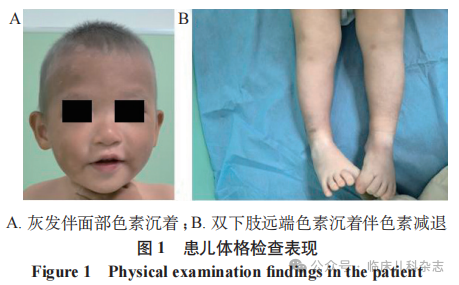
患儿,男,1 岁 7 个月,父母非近亲结婚,因「面部、四肢远端色素沉着,伴灰发 1 年余」为主诉就诊于复旦大学附属儿科医院皮肤科。患儿生长发育正常,体重 13 kg,查体可见面颈部及手背部褐色色素沉着,四肢远端褐色色素沉着伴色素减退,边界不清。头发、眉毛呈灰色,毛发密度正常。指甲、牙齿正常(图 1)。颌下淋巴结及肝脾未触及肿大,无皮肤黏膜出血及虹膜色素沉着,心脏及神经系统查体均未见明显异常。家属诉该患儿生后多次上呼吸道感染病史,无相关家族史。
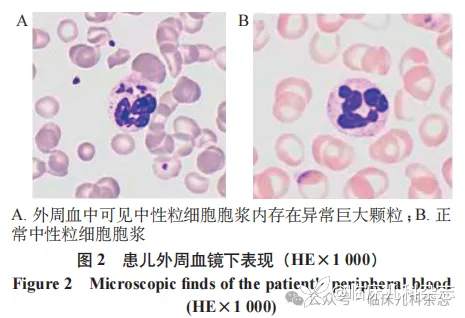
辅助检查:血常规及凝血功能未见明显异常。腹部超声未见肝脾肿大及占位。患儿外周血涂片发现部分中性粒细胞胞浆内颗粒增大、增多(图 2)。
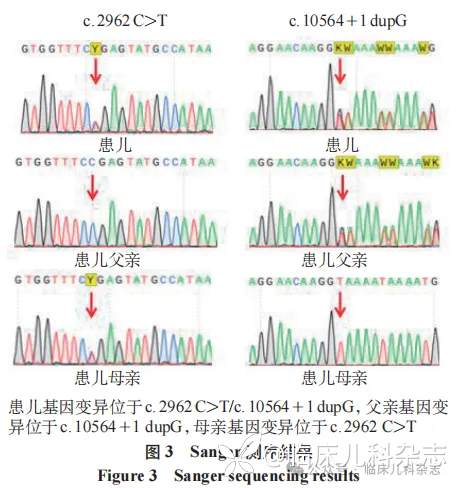
鉴于患儿病情,经家长签署知情同意书后应用全外显子组测序行相关遗传学检测。抽取患儿及父母外周血各 2 mL,进行家系全外显子组测序(trio whole-exome sequencing,trio-WES)及数据分析。检测发现患儿存在 LYST 基因的复合杂合变异。变异(NM_000081:exon6:c.2962C>T)为无义变异,可导致蛋白质翻译提前终止,此变异位点源自患儿母亲(图 3)。文献报道有 CHS 病例检测到该变异 [5]。另一个变异为剪接位点变异,变异(NM_ 000081 :intron46:c.10564+1dupG)影响 LYST 基因的转录,导致转录产物 mRNA 序列异常,引起 LYST 蛋白结构异常,此变异位点源自患儿父亲(图 3)。根据 ACMG 指南,该变异建议归类为致病(P)变异。多个剪接预测软件预测该变异可能导致异常剪接进而使该基因编码的蛋白产物功能发生改变,且已知该变异的下游多个截短变异被报道为致病/疑似致病变异;该变异未被 gnomAD(v4.1.0)公共数据库收录,为罕见变异。
2 讨论
CHS 是一种罕见的常染色体隐性遗传病,其特征包括部分性眼皮肤白化病、出血倾向、免疫功能障碍及神经系统损害 [4,6]。其色素表现为涵盖皮肤、眼睛和毛发的色素稀释。值得注意的是,患者的头发可呈银灰色或带有金属光泽,这一特征在 CHS 动物模型中亦常被观察到。在某些人群中可出现虹膜 [6] 和皮肤 [7] 的过度色素沉着,尤其在日光暴露部位。国内报道的 CHS 患儿皮肤表现以弥漫性或局限性色素减退为主,表现为局部皮肤白化、毛发灰稀、虹膜色素减少等。而本例患儿的特点在于:四肢远端同时存在色素减退与色素沉着,这一混合性色素改变在既往国内文献中未见明确报道,为本病例的特殊临床表现之一 [8]。出血倾向通常较轻微,表现为易出现瘀伤和黏膜出血。免疫功能障碍则表现为频繁复发感染,常见于皮肤和上呼吸道。大多数患者从人生的第二个十年开始出现神经系统并发症,可包括认知障碍、感觉和运动神经病变、共济失调、痉挛性截瘫及帕金森综合征 [7,9-10]。近期研究发现,肌萎缩侧索硬化症(ALS)[9] 和噬血细胞性淋巴组织细胞增多症(HLH)[10] 与 LYST 基因的双等位基因错义变异存在关联,且不伴具有病理诊断意义的胞浆内巨大颗粒;这些发现是否代表了 CHS 临床谱系的扩展,尚需进一步研究来确认。
LYST 基因含有 53 个外显子,其中 51 个外显子编码蛋白。LYST 蛋白是 BEACH(Beige and Chediak Higashi)蛋白家族成员之一,BEACH 蛋白存在于所有真核生物中 [11]。LYST 蛋白主要含有 3 个区域:氨基端 ARM/HEAT 区、羧基端的 BEACH 结构域及其下游的 WD40 重复序列 [12]。其中 LYST 基因羧基末端区域(包含 BEACH 结构域及 WD40 结构域)对蛋白功能至关重要,该区域的错义或非移码插入/缺失可能导致保留部分的蛋白功能,从而表现为较轻的临床表型;而该区域的无义或移码变异则通常导致蛋白截短、功能完全丧失,与重型 CHS(儿童期即出现 HLH 及神经系统受累)相关。根据对已报道 CHS 病例文献的综合分析,携带至少一个错义或框内变异通常与较轻的临床表现相关,而携带两个无义或移码变异则多导致更严重的疾病表型 [13]。本例患儿虽携带一个无义变异和一个剪接位点变异,但尚未观察到出血倾向、免疫功能障碍及神经系统损害等典型严重表现。其一,可能与患儿年龄较小有关,此类严重并发症多出现在生命的第二个十年之后。其二,从遗传机制角度考虑,该剪接位点变异可能并未引起完全异常的蛋白质产物,而是仅导致部分外显子缺失或产生部分功能正常的剪接异构体。这些保留部分功能的蛋白仍具有一定程度的溶酶体运输调节能力,从而在一定程度上延缓或减轻了严重临床表现的出现。因此,尽管该患儿目前无神经系统受累表现及出血倾向,但仍需进行长期随访观察。
综上所述,本报道通过对 1 例灰发伴色素异常的患儿进行全外显子组测序,结合临床表型诊断其为 CHS,通过与既往文献相比较,发现其与报道的 CHS 表型有不符之处,LYST 基因新发剪接位点变异可能是其遗传学病因。截至目前,CHS 国内外报道较少,本案例发现的剪接位点变异未见报道,是一个新的变异,这扩充了 LYST 基因缺陷的基因型-表型谱,也为进一步研究 CHS 提供素材和参考。
好文章,需要你的鼓励