



来源:《临床儿科杂志》
胡佳悦, 应令雯, 常国营, 李娟, 杨帆, 王翠锦, 郁婷婷, 姚如恩, 罗成娟, 王秀敏. 8 例儿童异染性脑白质营养不良临床表型、遗传学分析及异基因造血干细胞移植疗效观察 [J]. 临床儿科杂志, 2025, 43(10): 734-741 DOI:10.12372/jcp.2025.25e0117
HU Jiayue, YING Lingwen, CHANG Guoying, LI Juan, YANG Fan, WANG Cuijin, YU Tingting, YAO Ruen, LUO Chengjuan, Wang Xiumin. Clinical phenotypes, genetic analysis and allogeneic hematopoietic stem cell transplantation efficacy of 8 children with metachromatic leukodystrophy[J]. Journal of Clinical Pediatrics, 2025, 43(10): 734-741 DOI:10.12372/jcp.2025.25e0117
本文作者: 胡佳悦 1 应令雯 2,3 常国营 1,2,3 李 娟 2,3 杨 帆 1 王翠锦 4 郁婷婷 3,5 姚如恩 3,5 罗成娟 6 王秀敏 1,2,3
作者单位: 上海交通大学医学院附属上海儿童医学中心 1. 临床研究病区,2. 内分泌遗传代谢科, 3. 上海市罕见病临床研究中心,4. 神经内科,5. 遗传分子诊断科,6. 造血干细胞移植科(上海 200127)
摘要:目的 分析 8 例异染性脑白质营养不良(MLD)患儿的临床特征和基因变异,探讨了基因型与临床表型的相关性及异基因造血干细胞移植(allo-HSCT)的疗效。方法 研究收集了 2013 年至 2024 年的患儿资料,通过全外显子组测序确诊,发现所有患儿均有发育迟缓等表现,确诊年龄 1 岁 3 个月至 9 岁 6 个月。结果 根据起病年龄和临床表现,4 例为晚婴型,2 例死亡;4 例为青少年型,生存率 100%。基因测序显示 ARSA 基因的复合杂合变异,共 15 种突变,其中 3 种为首次报道,均为有害变异。3 例患儿接受了 allo-HSCT 治疗,均存活但症状有进展。结论 MLD 主要表现为中枢神经系统损害,需结合临床表现、ARSA 酶活性及基因检测确诊。早期诊断和治疗对改善预后至关重要,allo-HSCT 可提高生存率,但疗效有限。
关键词:异染性脑白质营养不良;ARSA 基因; 溶酶体贮积病; 异基因造血干细胞移植
异染性脑白质营养不良症(metachromatic leukodystrophy,MLD ;OMIM 250100)是一种罕见的遗传性溶酶体贮积病,预估的出生患病率为 (1.4~1.8)/ 10 万,呈常染色体隐性遗传 [1]。本病主要由编码溶酶体芳基硫酸酯酶 A(aryIsulfatase A,ARSA)的 ARSA 突变引起,少数由编码神经鞘脂激活蛋白 B(sphingolipid activator protein B,SAP-B,saposin B)的 PSAP 突变引起。ARSA 或 SAPB 蛋白的缺陷可导致溶酶体内硫酸脑苷脂水解受阻,并沉积在中枢神经系统的白质、周围神经及其他内脏组织中。无法水解的硫酸脑苷脂会聚集在神经元和生成髓鞘的细胞中,抑制髓鞘的形成,促进脱髓鞘的进展,引起脑白质、周围神经及其他内脏组织等病变,从而产生严重代谢异常的脱髓鞘性神经系统退行性疾病 [2-5]。
根据发病年龄不同,MLD 分为晚婴型(2.5 岁前发病)、青少年型(2.5~16 岁发病)和成人型(16 岁后发病)[6-7]。其中,快速进展的运动障碍是晚婴型和青少年型的特征。认知和行为障碍是成人型的特征。既往研究表明,MLD 的临床表现、残存的酶活性与基因突变位点密切相关,即酶活性越低,发病越早、病情越重:晚婴型通常为两个无效等位基因突变,无法表达任何有活性的 ARSA 蛋白,导致疾病快速进展;青少年型多由一个无效等位基因突变和一个可少量表达 ARSA 蛋白的基因突变;成人型多为 2 个可少量表达 ARSA 蛋白的双等位基因突变,其体内 ARSA 活性为正常人的 2%~4%[8-11]。截至目前,已有超过 200 余个 ARSA 变异被报道(Human Gene Mutation Database),相关临床表型严重程度不一。
目前,MLD 尚缺乏特异性根治疗法,临床管理以对症治疗为主 [6]。异基因造血干细胞移植(allo-HSCT)作为一种侵入性探索治疗手段已在部分病例中应用,临床观察表明对于症状前或极早期接受移植的患者,可能延缓甚至部分阻断疾病进程,但该疗法存在较高风险,临床数据显示对晚婴型患者疗效有限,即使在青少年型及成人型病例中亦存在显著个体差异 [12]。近来,基因治疗 [13-14] 和鞘内注射酶替代疗法 [15] 作为 MLD 的潜在治疗方法正逐步开展,然而,需要进一步的研究来评估它们的长期安全性和有效性。
尽管中国 MLD 患儿的基因型-表型关联分析已有初步研究 [16-17],但中国患儿群体进行 allo-HSCT 的风险效益及长期预后评估仍缺乏相关数据。据此,本研究回顾性分析了 2013 年 8 月至 2024 年 11 月在本中心诊疗的 MLD 患儿的临床特征、基因变异谱及长期治疗结局,以期通过总结 MLD 的临床、遗传学特征及 allo-HSCT 的风险疗效分析,为中国 MLD 患儿的早期诊断和治疗提供依据。
1 对象与方法
1.1 研究对象
选取 2013 年 8 月至 2024 年 11 月于上海儿童医学中心就诊的 MLD 患儿,并收集相关临床信息。其中,经筛选后共计 8 例患儿(P1~P8)被纳入本次研究。
MLD 诊断参考美国异染性脑白质营养不良监测和管理的共识指南 [13]:①临床表现:晚婴型多发生发育迟缓,失去语言能力,行走障碍;青少年型最初征兆可能是行为问题以及学习障碍;②检测血白细胞及皮肤成纤维细胞中 ARSA 活性低于正常对照的 10%;③尿硫酸脑苷脂检测阳性,尿沉渣发现大量异染颗粒;④头颅 MRI 通常提示脑白质区有高亮区域,脑室周围及皮质下白质广泛的、对称性的改变(说明髓鞘丢失),或提示脑白质营养不良;⑤基因检测显示 ARSA 致病变异。所有研究对象家长或监护人均签署知情同意书,本研究经医院医学伦理委员会的审查通过(No.SCMCIRB-K 2020060-1)。
1.2 研究方法
1.2.1 临床资料收集 采集包括患儿病史、家族史、颅脑 MRI 以及脑电图结果,患儿及其家系成员基因检测结果等信息。
1.2.2 测定 ARSA 酶活性 通过密度梯度离心法从患儿外周血中分离白细胞,4℃ 下经超声破碎裂解 3 次,置于 0℃ 孵育 17 小时后,加入氢氧化钠终止反应。使用对硝基儿茶酚硫酸盐作为底物,通过分光光度计检测 515nm 处的吸光度,结合蛋白质浓度计算 ARSA 酶活性 [18]。正常值范围为 218~ 550nmol·17 h-1·mg-1,低于此阈值提示 ARSA 活性低下,结合临床表型与基因检测可辅助诊断 MLD。
1.2.3 全外显子组测序 基因检测和变异评估采集患儿和父母外周血,使用 Gentra Puregene Blood Kit(Qiagen)从外周血白细胞中分离基因组 DNA。采用安捷伦 SureSelect 外显子捕获法和 Illumina 测序平台(美国 Illumina 公司)进行全外显子组测序。测序数据质控后经 BWA 软件比对、GATK 软件检测变异后,用 Flash Analysis 在线软件系统进行变异过筛及解释,候选变异经 Sanger 测序验证,同时通过比较样本区间内测序 reads 数目判断全基因组拷贝数。
1.2.4 致病性分析 在 ClinVar(https://www. ncbi.nlm.nih.gov/clinvar)和 HGMD(https://www. hgmd.cf.ac.uk/ac/index.php)数据库中搜索正常人群中候选变异的携带频率。利用 PolyPhenG2、SIFT 和 Mutation Taster 等在线软件预测候选变异对编码蛋白功能的影响。使用 alphafold2 软件预测变异蛋白的结构,使用 AlphaMissense 预测突变氨基酸致病性。结合美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)和临床基因组资源中心(Clinical Genome Resource,ClinGen)联合颁布的指南对位点进行致病性评估 [19-20],并将其分为良性、可能良性、意义不明确、可能致病性与致病性。
1.2.5 Sanger 测序验证 设计针对突变位点的 PCR 扩增引物,采用 Taq DNA 聚合酶试剂盒对 DNA 样本扩增,扩增后电泳目标条带特异,使用 ABI3500 测序仪进行 Sanger 测序验证。
2 结果
2.1 临床分析及 allo-HSCT 治疗结果
共有 8 例 MLD 纳入本次研究,其中男性 4 例,女性 4 例,中位起病年龄 4.3 岁,中位确诊年龄 4.7 岁,起病至确诊时间间隔约 5 个月,见表 1。进一步根据起病年龄进行分型可见,4 例为晚婴型(男性 3 例,女性 1 例),均以行走困难起病,起病年龄 9 月龄至 2 岁 6 个月,中位起病年龄为 2.1 岁,中位确诊年龄 2.5 岁,起病至确诊时间间隔约 5 个月。另有 4 例为青少年型,主要表现为智力落后及运动倒退,起病年龄在 6 岁至 9 岁 1 个月,中位起病年龄 6.5 岁,确诊年龄 7.2 岁,起病至确诊时间间隔约 9 个月,相较晚婴型确诊时间有所延长。
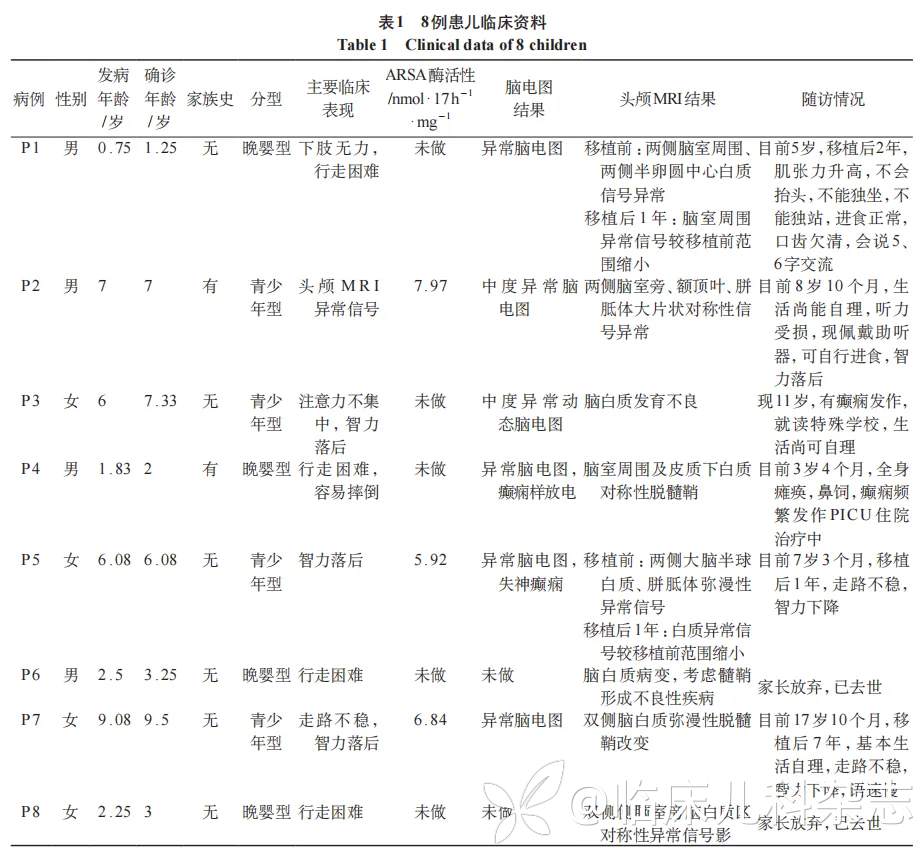
辅助检查结果显示,所有 MLD 患儿均存在 ARSA 酶活性的显著降低,并在起病时或疾病进程中出现中枢神经系统累及。其中,P2~P4、P6 和 P8 头颅磁共振成像(MRI)结果提示脑白质发育不良;P1、P5 和 P7 头颅 MRI 结果提示双侧脑白质弥漫性脱髓鞘改变;P3 脑电图提示中度异常动态脑电图;P5 脑电图提示异常脑电图,失神癫痫。此外,P2 还表现为听力落后,予助听器辅助。
共有 3 例患儿进行了异基因造血干细胞移植(allo-HSCT)治疗,截止目前,最长随访时间为移植后 7 年,所有患儿均存活,且未出现移植物抗宿主病、移植后淋巴增殖性疾病等移植相关并发症。
对于晚婴型患儿,1 例患儿(P1)进行了 allo-HSCT,目前 5 岁,移植后 2 年,可正常进食,但临床症状仍较前加重,表现为肌张力增高及运动语言发育迟缓(不会抬头、不能独坐、不能独站,口齿欠清,仅能 5~6 字交流)。而对于未进行 allo-HSCT 治疗的 3 例晚婴型患儿,P4 目前 3 岁 4 个月,全身瘫痪,鼻饲维持中,伴癫痫频繁发作,PICU 住院治疗中;另 2 例患儿(P6 和 P8)因病情危重,家长放弃治疗后死亡。
对于青少年型患儿,2 例患儿(P5 和 P7)进行了 allo-HSCT,临床症状较前稍有进展。其中,P5 患儿目前 7 岁 3 个月,移植后 1 年,较前出现走路不稳伴智力下降;P7 患儿目前 17 岁 10 个月,移植后 7 年,仍有走路不稳,智力较前下降,讲话语速慢,但基本生活可自理。对于未进行 allo-HSCT 治疗的 2 例青少年型患儿,尽管生活尚可自理,但均表现出不同程度的疾病进展。P2 患儿目前 8 岁 10 个月,表现为智力落后,伴听力受损,目前助听器辅助;P3 患儿现 11 岁,就读于特殊学校,伴癫痫发作。
2.2 遗传学分析结果
本研究纳入的 8 例患儿来自 8 个不同的家系,其中 2 例患儿存在阳性家族史,见图 1。P2 患儿有 2 位胞姐确诊 MLD,均 9 岁起病,现一位走路易摔倒,另一位瘫痪在床,二便失禁。P4 患儿有 1 妹妹目前 1 岁,通过基因检测确诊 MLD,暂无临床表现。
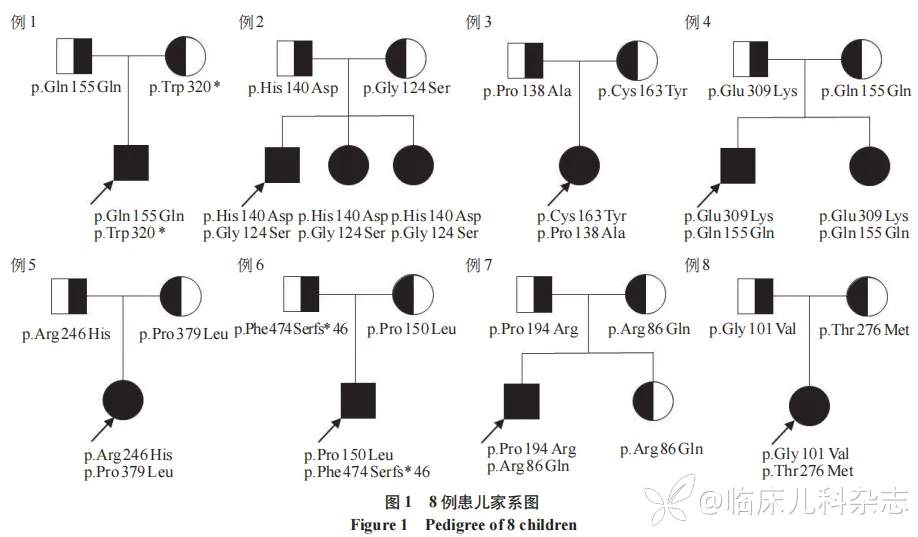
进一步的全外显子组测序结果显示,8 例患儿均携带 ARSA 复合杂合变异,共检测到 15 种基因突变,86.7%(13/15)为错义突变,13.3%(2/15)为无义突变,所有突变均来源于亲代。其中,P3 和 P6 分别携带的 3 种突变形式 c.412C>G(p.Pro138Ala)、c.488 G>A(p.Cys163Tyr)和 c.1421delT(p.Phe474Serfs*46)既往未被报道。见表 2,图 1。
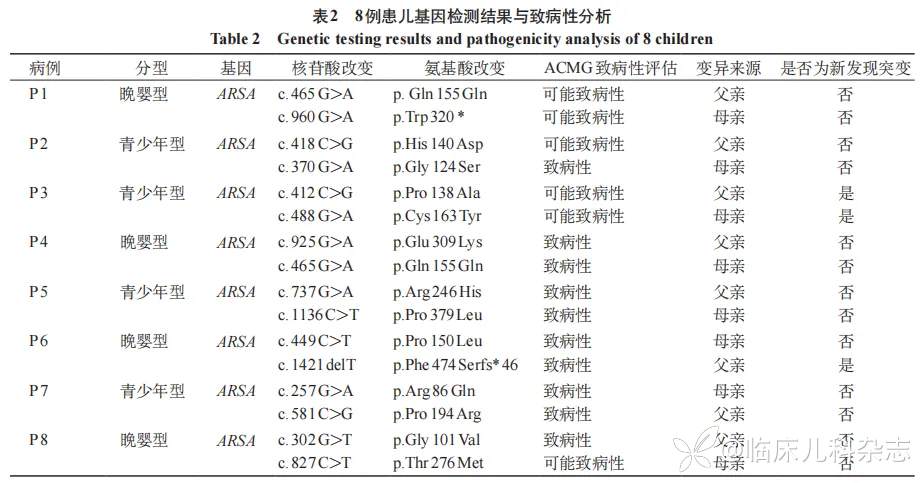
通过 Sanger 测序验证了 ARSA 的错义变异 c.412C>G 导致的 p.Pro138Ala 未在 HGMD 和 ClinVar 等数据库中发现(PM2),且有其他致病性变异在同一位置的病例报告(PM5)[21-24]。P3 患儿的临床表型高度符合 MLD(PP4),在线软件 PolyPhenG 2、SIFT 和 Mutation Taster 的预测表明,该变异可能会影响蛋白质结构域的功能(PP3)。根据 ACMG 指南,该变异被评定为可能致病性变异(PM2+PM5+PP4+PP3)。使用 alphafold2 预测突变蛋白结构并与野生型蛋白(P15289)进行比较,发现突变氨基酸引起二级结构改变。脯氨酸(P)具有刚性环状结构,在蛋白质折叠中起到维持结构稳定性和约束局部结构的作用。脯氨酸在蛋白质二级结构中常常作为转折点或改变折叠方向的关键残基,将 P 替换为丙氨酸(A),环状结构缺失,可能会导致蛋白质折叠的改变,尤其是在该位点附近的二级结构变化。使用 AlphaMissense 预测突变氨基酸致病性发现该氨基酸错义变异预测为有害性(AlphaMissense pathogenicity score: 0. 944)。
P3 患儿的另一个错义变异 c.488 G>A 导致的 p.Cys163Tyr 未在 HGMD 和 ClinVar 等数据库中发现(PM2),且该患儿的另一复合杂合变异经上述判断为可能致病性变异(PM3)。P3 患儿的临床表型同样高度符合 MLD(PP4),在线软件 PolyPhenG2、SIFT 和 Mutation Taster 的预测表明,该变异可能会影响蛋白质结构域的功能(PP3)。根据 ACMG 指南,该变异被评定为可能致病性变异(PM2+PM3+PP4+PP3)。使用 alphafold2 预测突变蛋白结构并与野生型蛋白(P15289)进行比较。C163 位与临近氨基酸形成二硫键,维持蛋白三维结构和稳定性,半胱氨酸(C)突变为酪氨酸(Y),原二硫键结构遭到破坏,可能影响蛋白结构和稳定性。使用 AlphaMissense 预测突变氨基酸致病性发现该氨基酸错义突变预测为有害性(AlphaMissense pathogenicity score: 0.977)。
P6 患儿的 c.1421delT 导致的移码突变 p.Phe474Serfs*46 会过早产生一个终止的密码子,并通过无义介导的 mRNA 衰变显著损害 ARSA 活性,为无效等位基因突变,根据 ACMG 指南,该变异被评定为致病性变异(PVS1+PM2+PM3)。使用 alphafold2 预测突变蛋白结构并与野生型蛋白(P15289)进行比较。在突变蛋白中,第 474 位的苯丙氨酸(F)被丝氨酸(S)替代,并且该突变导致框移,进而改变了后续氨基酸的翻译序列和蛋白结构。
3 讨论
MLD 是一种罕见遗传代谢病,具体患病率未知,且具有种族差异。在美国,MLD 的发病率约为 1/40000,估计每年有 3600 例 MLD 新生儿出生 [25]。对于中国人群,据中国医药信息查询平台显示,MLD 的发病率为 (0.8~2.5)/ 10 万人,其中婴儿晚发型最为多见,占全部病例的 60%~70%,青少年型次之,约占 20%~30%。本研究纳入的 8 例 MLD 患儿均有典型的临床症状,最终经 ARSA 酶活性、全外显子组测序结果验证确诊为 MLD,其中晚婴型患儿 4 例(50%),青少年型患儿 4 例(50%)。与既往研究一致,4 例晚婴型患儿均以行走困难、容易摔倒起病,病程进展迅速,预后不佳 [26-27]。2 年随访时,2 例患儿已死亡,存活的 2 例患儿均表现为明显的中枢神经障碍。4 例青少年型患儿均因智力落后来院就诊,截止末次随访,智力进一步下降,同时因合并共济失调和轻微的锥体综合征,表现为步态问题。对于该类患儿,尽管短期内疾病进展比晚婴型缓慢,但疾病后期病情会迅速进展,痉挛变得突出,发展为癫痫。在青少年 MLD 患者中,头颅 MRI 特征性白质病变在相关症状出现时就已经存在,且其进展模式的异质性显著高于晚婴型患儿。
MLD 通常由 ARSA 变异导致的 ARSA 缺乏所致。迄今,根据人类基因突变数据库报道,全球共有 200 余个 ARSA 变异以错义/无义突变最为常见,缺失突变次之。与既往的报道一致,本研究纳入的患儿均携带 ARSA 的复合杂合突变。15 种突变类型中,86.7% 为错义突变,13.3% 为无义突变。此外,本研究中 3 种突变既往未被报道,经 ACMG 评估后发现,c.488 G>A(p.Cys163Tyr)为可能致病性变异(PM2+PM3+PP4+PP3),c.412C>G(p.Pro138Ala)为可能致病性变异(PM2+PM5+PP4+PP3),c.1421delT(p.Phe474Serfs*46)为致病性变异(PVS1+PM2 +PM3)。与此同时,我们的基因型 -表型分析结果确认了 8 种无效等位基因突变,与既往报道结果一致 [16,28]。结果显示,P1、P4、P6 和 P8 携带的 c.302 G>T(p.Gly101Val)、c. 449C>T(p.Pro150Leu)、c.465 G>A(p.Gln 155 Gln)、c.737 G>A(p.Arg246 His)、c.827C>T(p.Thr276Met)、c.925 G>A(p.Glu309Lys)、c.960 G>A(p.Trp320*)和 c.1421delT(p.Phe474Serfs*46)是无效等位基因突变,与之更重的临床表型一致。除此之外,因 P138 是人类其他几种硫脂酶家族以及其他物种的硫脂酶中进化上保守的氨基酸残基之一,对这一氨基酸残基的替换可能会导致蛋白质折叠的改变,尤其是在该位点附近的二级结构变化,并与严重的表型相关。P5 青少年型患儿的 p.Pro379Leu 突变在既往报道中显示残存约 12.1% 的 ARSA 活性,被归类为轻微突变 [24],另一突变 p.Arg246 His 则考虑为无效基因突变。
allo-HSCT 是目前治疗 MLD 的主要方法,然而该疗法的有效性仍存在争议。一般来说,allo-HSCT 对有明显神经系统症状或晚婴型 MLD 患儿没有明显获益;而对于无症状、以及早期青少年 /成人型 MLD 患儿来说,allo-HSCT 可以延缓疾病进展并改善预后。据报道,移植个体对大运动或语言功能的影响较未移植个体小,且长期随访中可见到部分髓鞘再生 [6]。而在本研究中,我们发现婴儿晚期型患儿的生存期较未移植的患儿延长,但由于疾病早期的损失,因此移植后的获益有限。青少年型患儿在移植后 1 年复查头颅 MRI 检查中发现,患儿异常信号较移植前范围缩小,但临床症状改善有限,尚需长期的随访以进一步观察治疗效果及预后。在本研究中,基因型与结局之间没有明确的关联。
近年来,随着基因治疗的迅猛发展,Atidarsagene autotemcel 于 2020 年 12 月被欧盟批准用于 MLD 的治疗,并在今年 3 月获得美国 FDA 批准 [13-14]。然而,该疗法仅被批准用于无症状晚婴型 MLD、无症状/早期青少型 MLD 的个体,同时考虑到其高昂的治疗成本,allo-HSCT 仍是目前治疗 MLD 的主流方法。
本研究也存在局限性,回顾性设计导致部分数据缺失,且样本量较小(尤其 allo-HSCT 组仅 3 例),相关发现亟需有待前瞻性多中心研究加以验证。
综上,本文总结了 8 例 MLD 患儿的典型临床表型、遗传分析及治疗结局,进一步丰富了 MLD 的基因谱,为疾病的早期诊疗提供了依据。此外,allo-HSCT 可以改善 MLD 患儿的病情,提高晚婴型的存活率,但疗效尚有限,为中国 MLD 患儿的个体化治疗提供了新的参考依据。
好文章,需要你的鼓励