



来源:《临床儿科杂志》
裴培, 李伟华, 淮婉, 姚如恩, 葛禾佳, 王纪文, 王秀敏, 吉炜, 周昀箐, 贺影忠, 韩凤. ATP1A2 / ATP1A3 基因变异患儿 10 例遗传学及临床特征分析 [J]. 临床儿科杂志, 2025, 43(8): 590-597 DOI:10.12372/jcp.2025.24e1372
PEI Pei, LI Weihua, HUAI Wan, YAO Ruen, GE Hejia, WANG Jiwen, WANG Xiumin, JI Wei, ZHOU Yunqing, HE Yingzhong, HAN Feng. Genetic and clinical characteristics analysis of 10 children with ATP1A2/ATP1A3 gene variants[J]. Journal of Clinical Pediatrics, 2025, 43(8): 590-597 DOI:10.12372/ jcp.2025.24e1372
本文作者: 裴 培 1 李伟华 2 淮 婉 3 姚如恩 3 葛禾佳 1 王纪文 4 王秀敏 5 吉 炜 6 周昀箐 4 贺影忠 4 韩 凤 4
作者单位:1. 嘉兴大学附属第二医院儿童医学中心儿科(浙江嘉兴 314000);2. 上海交通大学医学院附属上海儿童医学中心儿科(上海 200127);3. 上海交通大学医学院附属上海儿童医学中心遗传分子诊断科(上海 200127);4. 上海交通大学医学院附属上海儿童医学中心神经内科(上海 200127);5. 上海交通大学医学院附属上海儿童医学中心内分泌代谢科(上海 200127);6. 上海交通大学医学院附属上海儿童医学中心心血管科(上海 200127)
摘要:目的 总结 10 例 ATP1A2 及 ATP1A3 基因变异患儿的临床表现和基因变异类型,丰富对该疾病的基因型-表型关联认识,促进临床医师对该疾病的了解。方法 回顾性收集 2015 年 11 月至 2024 年 6 月诊治的 10 例 ATP1A2 及 ATP1A3 基因变异患儿的临床资料,分析其遗传学及临床特征,结合文献检索,总结已报道的 ATP1A2 及 ATP1A3 基因变异类型与临床表现的关系。结果 共纳入 ATP1A2 及 ATP1A3 基因变异患儿 10 例,女 3 例、男 7 例,起病年龄 0 至 14 岁;以惊厥起病 3 例,以肌无力起病 2 例,偏瘫起病 1 例,交替性偏瘫起病 1 例,注意力缺陷障碍伴遗尿起病 1 例,1 例以惊厥合并肌无力起病,语言运动障碍起病 1 例。基因检测 ATP1A2 变异 3 例,ATP1A3 变异 7 例,均为点突变。目前 ATP1A2 基因(NM_000702.3)变异 c.587 G>A(p.Arg196 His)及 c.2841-2A>C 突变位点和 ATP1A3 基因(NM_152296.4)变异 c.1013C>T(p.Ala338Val)及 c.2426C>A(p.Ala809Asp)突变位点尚未见报道。结论 新发现的变异丰富了 ATP1A2 及 ATP1A3 基因变异谱及临床表现谱,同时提示临床工作者对于以癫痫、肌无力及偏瘫等神经症状为首发表现的疾病,若疗效欠佳时应及时完善基因检测以明确诊断。
关键词: ATP1A2 基因; ATP1A3 基因; 儿童交替性偏瘫; 家族性偏瘫性偏头痛; 癫痫
钠/钾转运 ATP 酶α2 亚基(ATPase,Na/K transporting Alpha 2 polypeptide,ATP1A2)基因和钠/钾转运 ATP 酶α3 亚基(ATPase,Na/K transporting Alpha 3 polypeptide,ATP1A3)基因分别编码 Na+/K+-ATP 酶(Na+/K+-ATPase,NKA)离子泵的α2 和α3 亚单位,这些亚单位在维持细胞内外钠、钾离子浓度平衡方面发挥着关键作用 [1]。ATP1A2 基因于中枢神经系统中广泛表达,在大脑皮层、海马体等区域的表达水平较高 [2],参与调节神经元的电生理特性,而 ATP1A3 基因主要在γ-氨基丁酸(GABA)能神经元中表达 [3],但两种基因变异在临床表现上均与家族性偏头痛、癫痫及其他神经系统疾病密切相关 [4-6]。尽管已有研究报道 ATP1A2 和 ATP1A3 基因的多种变异类型及其临床表现,但研究集中于特定的变异类型,缺乏对这些变异类型与临床症状差异的系统性总结及对不同变异类型在临床表现和预后方面的全面性比较。这种局限性不仅影响对患儿的准确诊断,也限制了对潜在治疗策略的探索。
因 此 ,本研究旨在总结收治的 ATP1A2 和 ATP1A3 基因变异患儿的临床表现及基因特征,通过对这些病例的深入分析,并结合已发表的相关文献,为临床医师提供更为全面的 ATP1A2 和 ATP1A3 基因变异相关知识,以促进对相关疾病的早期诊断和个体化治疗。
1 对象与方法
1.1 研究对象
本研究通过病历系统检索 2015 年 11 月至 2024 年 6 月在嘉兴大学附属第二医院儿童医学中心和上海交通大学医学院附属上海儿童医学中心诊治的 ATP1A2/ATP1A3 基因变异的相关患儿 10 例。纳入标准 :①明确基因检测为 ATP1A2/ATP1A3 基因变异,且为致病性或可能致病性变异;②有癫痫、肌无力及偏瘫等典型的临床表型;③年龄<18 岁。排除标准:临床资料不全。
1.2 方法
1.2.1 临床资料收集 从病历系统中采集以下信息:性别、起病年龄、确诊年龄、首发表现、脑电图、头颅影像、发育情况、基因检测相关数据(如基因类型、突变类型、突变位点、氨基酸改变、家族情况),变异依据美国医学遗传学与基因组学学会(ACMG)评级。
1.2.2 基因检测 利用 Agilent SureSelect 方法外显子捕获(ExomeV6),Illumina 测序平台进行高通量测序,测序数据经 NextGENe®软件匹配分析后,用 Ingenuity 在线软件系统进行变异过筛及解释,候选变异经 Sanger 测序验证。
1.2.3 文献检索 分别以「ATP1A2」、「ATP1A3」为关键词,对中国科学期刊全文数据库、万方数据知识服务平台和 NCBI、PubMed 等数据库截止至 2024 年 6 月所有收录的论文进行检索,查找相关基因变异的文献报道,收集已公开发表的期刊论文、书籍、会议论文及学位论文;患者均通过基因检测发现明确的 ATP1A2/ATP1A3 基因致病性变异。
2 结果
2.1 基本临床资料
共收集 10 例 ATP1A2/ATP1A3 基因变异患儿,女 3 例、男 7 例,起病年龄为 0 至 14 岁 ;以惊厥起病 3 例,以肌无力起病 2 例,偏瘫起病 1 例,交替性偏瘫起病 1 例,注意力缺陷障碍伴遗尿起病 1 例,以惊厥合并肌无力起病 1 例,语言运动障碍起病 1 例。实验室检查提示,5 例脑电图出现异常,均提示慢活动增多 ;3 例头颅磁共振成像(MRI)有异常,2 例表现为脑沟变深变宽,1 例侧脑室形态不对称。随访 9 例患儿有语言运动落后,1 例发育正常。见表 1。

2.2 基因检测结果
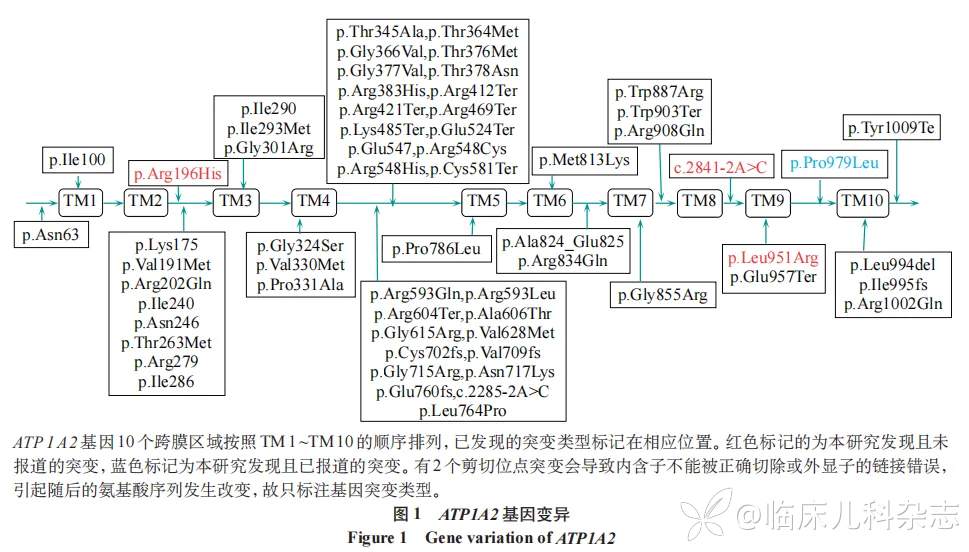
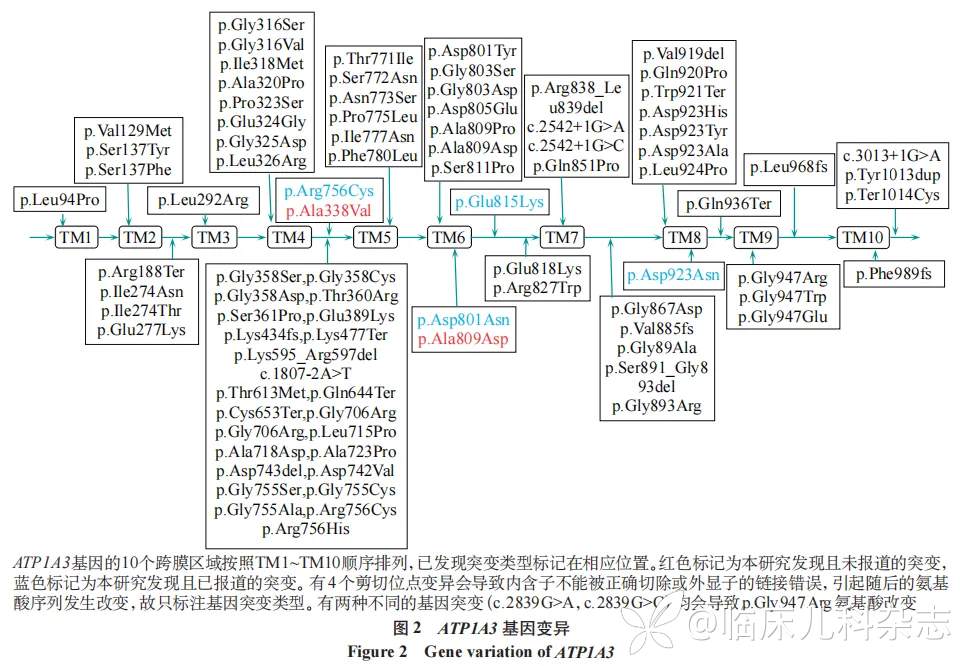
基因测序结果显示 ,ATP1A2 变异 3 例 ,ATP1A3 变异 7 例,均为点突变,具体详见表 1、表 2。目前 ATP1A2 基因 (NM_000702.3 ) c.587 G>A(p.Arg 196 His)及 c.2841-2A>C 突变位点和 ATP1A3 基因 (NM_152296.4 ) c.1013C>T(p.Ala338Val)及 c.2426C>A(p.Ala809Asp)突变位点尚未见报道。

2.3 ATP1A2/ATP1A3基因检索结果
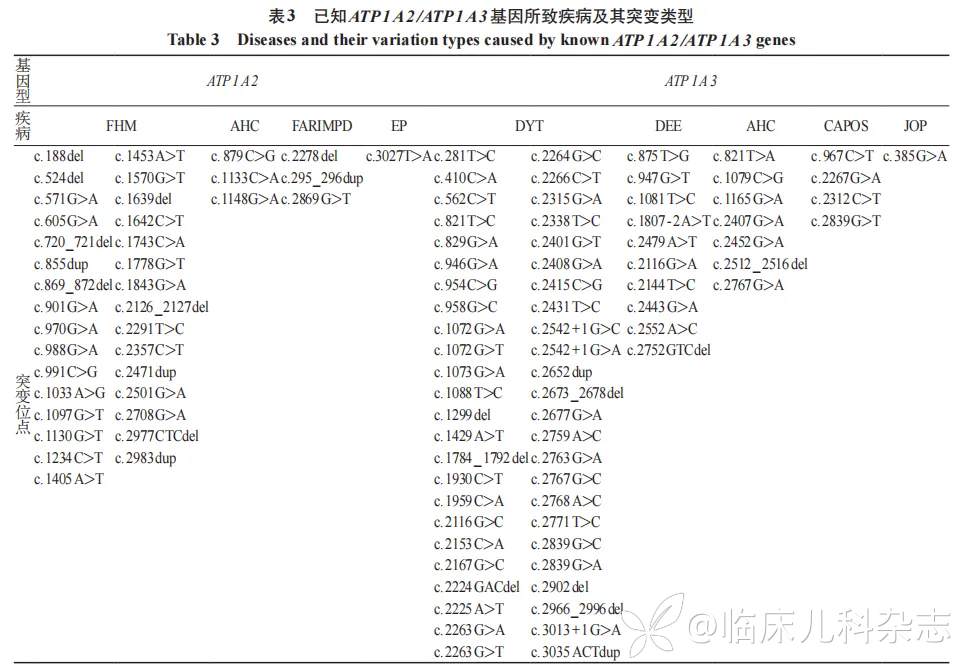
根据参考条件,共检索到 59 例致病性 ATP1A2 基因变异,44 例错义突变,11 种缺失突变,4 例重复突变。ATP1A2 基因序列中的突变位点见图 1,其中有 34 例突变临床表现为家族性偏瘫性偏头痛(familial hemiplegic migraine,FHM),3 例突变表现为儿童交替性偏瘫(alternating hemiplegia of childhood,AHC),3 例突变表现为胎儿运动不能、呼吸功能不全、小头畸形、多小脑回和畸形面容综合征(fetal akinesia,respiratory insufficiency,microcephaly,polymicrogyria and dysmorphic facies,FARIMPD),1 例突变表现为癫痫(epilepsy,EP),余突变类型未提供相关临床表现。同时检索到 86 例致病性 ATP1A3 基因变异,75 例错义突变,9 例缺失突变,2 例重复突变。ATP1A3 基因序列突变位点见图 2,其中有 48 例突变表现为肌张力障碍(dystonia,DYT),10 例突变表现为发育性癫痫性脑病(developmental and epileptic encephalopathy,DEE),7 例突变表现为 AHC,4 例突变表现为小脑性共济失调、反射消失、弓形足、视神经萎缩和感音神经性听力损失综合征(cerebellar ataxia, areflexia, pes cavus, optic atrophy, and sensorineural hearing loss,CAPOS),1 例突变表现为青少年精神病(juvenileonset psychosis,JOP),余突变类型未提供相关临床表现。见表 3。
3 讨论
NKA 离子泵是一种位于细胞质膜上普遍存在的跨膜离子泵,负责 Na+-K+离子的主动交换 [7]。神经组织中 NKA 还可以通过协调细胞膜上的离子浓度梯度参与 Ca2+信号转导和神经递质释放,以维持神经细胞上的静息电位、兴奋电位和主动转运 [8]。NKA 是一种异源三聚体α-β-γ蛋白复合物,由一个大的α亚基和较小的β和γ亚基组成 [9]。目前已知有四种α亚型(α1、α2、α3 和α4),分别由四个旁系同源基因(ATP1A1、ATP1A2、ATP1A3、ATP1A4)编码,此 4 种基因共享 84%~91% 的氨基酸序列,并且具有发育和组织表达特异性 [1]。ATP1A1 和 ATP1A4 基因变异导致的神经系统症状较少,且临床表现轻,而 ATP1A2 和 ATP1A3 基因的结构性杂合突变通常与几种常染色体显性遗传神经系统疾病密切相关。
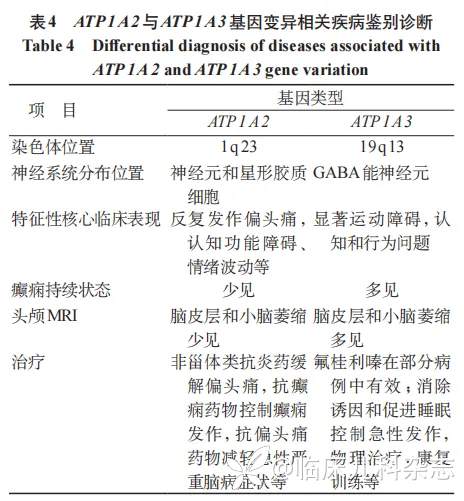
ATP1A2(OMIM# 182340)位于染色体 1q23 上,突变所致疾病主要通过常染色体显性遗传,多数家系呈现多代成员发病,有表型异质性特征;ATP1A3(OMIM#182350)位于染色体 19q13 上,突变所致疾病大多数为散发性,无家族史,少数以常染色体显性遗传为主,且同样具有表型异质性特征 [10]。两者在染色体上的位置有别,其调控机制及遗传连锁关系亦存在差异。ATP1A2 基因的变异会影响其编码的 NKA 酶α2 亚基的结构和功能,导致蛋白质折叠错误、稳定性下降或离子转运活性减弱,破坏细胞内外的离子平衡,进而影响神经元的兴奋性和神经信号传递。具体而言,此种失衡可能干扰神经递质的释放机制,影响突触传递的效率和准确性,进而引发一系列神经系统异常症状 [11]。ATP1A3 基因的变异会改变 NKA 酶的构象和离子结合位点,致使离子泵功能失调,破坏细胞内的离子稳态和信号转导通路,导致神经元能量代谢紊乱,还可能对神经胶质细胞功能产生影响,因此变异会妨碍细胞内能量物质的合成与利用,致使细胞能量供应不足,同时影响神经胶质细胞对神经元的营养支持和代谢调节功能 [12]。
自 2003 年和 2004 年 ATP1A2 和 ATP1A3 变异相继被报道,越来越多的变异类型和临床表现被人们发现。ATP1A2 基因变异的儿童,常见症状包括反复发作的偏头痛,其疼痛程度和发作频率各异。部分患儿的偏头痛可能呈现出周期性和剧烈性的特点,严重影响其日常生活和学习 [13]。ATP1A2 基因变异患儿还有认知功能障碍、情绪波动等神经精神症状 [14]。ATP1A3 基因变异患儿的临床表现多样,但共同特点是显著运动障碍 [15],包括 AHC、DYT 和 CAPOS 等,通过外显子测序发现典型 AHC 患者 95.7% 携带 ATP1A3 变异。ATP1A3 基因变异患儿同样有认知和行为问题,但与 ATP1A2 基因变异导致的情绪波动等神经精神症状不同,ATP1A3 基因变异多表现为注意力缺陷多动障碍(attention deficit and hyperactive disorder,ADHD)、自闭症谱系障碍(autism spectrum disorder,ASD)等,这些症状的出现可能与 ATP1A3 变异导致的神经元功能障碍有关,尤其是在大脑发育早期阶段 [16-17]。
本研究 3 例 ATP1A2 基因变异相关疾病中 ,DYT、AHC 及 DEE 各 1 例,7 例 ATP1A3 基因变异相关疾病中,DEE 4 例,AHC、DYT 各 1 例,DEE 合并 DYT 1 例。10 例患儿中,以惊厥起病 3 例,以惊厥合并肌无力起病 1 例,其中 1 例 ATP1A2 基因变异患儿出现惊厥持续状态,既往研究尚未见报道,考虑该基因变异引起跨膜蛋白结构改变,影响离子通道和泵的功能,进而影响神经元的兴奋性和稳定性,此发现可完善 ATP1A2/ATP1A3 基因变异相关疾病的临床表现谱。本研究 1 例患儿因生后肌无力诊断为脑瘫,予对症治疗效果欠佳,说明临床表现多样性增加了该类疾病的确诊难度。
文献检索目前报道了 59 例致病性 ATP1A2 基因变异和 86 例 ATP1A3 基因变异患者,本研究分析其遗传资料并对 ATP1A2/ATP1A3 基因变异的类型和频率进行评估,发现跨膜域 TM4 与 TM5 之间是突变热点。相关研究显示,由于α亚基细胞外环很短(TM7~TM8 环除外),主要由 10 个跨膜结构域(TM1~TM10)和 3 个胞内环组成,TM4 和 TM5 之间有大的中心环,由 430 个氨基酸残基组成,形成磷酸化(P);约 90 个氨基酸残基的长 N 末端尾,形成核苷酸结合(N);以及 TM2 和 TM3 之间有约 120 个残基的胞内环,形成致动器(A),这也导致突变位点更倾向于优先聚集在胞质区域,特别是 P 结构域 [18]。检索文献中 ATP1A2 基因 TM4 至 TM5 之间有 29 例突变,包括 25 例点突变,4 例缺失突变 ;ATP1A3 基因 TM4 至 TM5 之间有 26 例突变,包括 22 例点突变,4 例缺失突变。
已发表的 59 例 ATP1A2 基因变异中有 14 例氨基酸改变位于跨膜域;而已发表的 86 例 ATP1A3 基因变异中有 43 例氨基酸改变位于跨膜域。本研究对于这些疾病的基因突变类型进行了扩增,并对其首发症状进行补充。同种基因不同的突变类型会导致不同的临床表现,这种表型异质性意味着遗传和环境因素均影响 ATP1A2/ATP1A3 基因变异相关疾病的最终临床表现。
随着头颅 MRI 技术的不断发展,对于 ATP1A2 /ATP1A3 基因变异相关疾病患儿的脑组织结构研究也越来越多。ATP1A2/ATP1A3 基因变异会导致多种神经系统疾病,这些疾病的头颅影像提示大脑皮层和小脑的结构改变。有研究报道 ATP1A3 变异会引起早期严重的癫痫发作和小头畸形,以及反复发作性小脑共济失调和意识改变,此外,ATP1A3 基因变异可能引起大脑皮层发育异常,如多微小脑回症 [19-20]。本次 10 例患儿中,共 3 例患儿头颅影像异常,均为 ATP1A3 基因变异,考虑 ATP1A3 基因变异导致的神经损伤。
对于 ATP1A2 基因变异相关疾病的治疗主要是对症治疗,如使用非甾体类抗炎药(如美金刚)、曲坦类药物缓解偏头痛,使用抗癫痫药物控制癫痫发作等 [21-22],但对于重症患儿,预防谷氨酸兴奋毒性损伤和保护线粒体功能是早期治疗的重点,同时使用传统抗偏头痛的药物也可以有效减轻急性严重脑病的症状,并使其能在短时间内恢复,但治疗效果因人而异 [23]。而 ATP1A3 基因变异引起的疾病治疗更加复杂,多为支持性治疗,如物理治疗、康复训练,以提升运动功能和生活质量,一些药物如氟桂利嗪在部分病例中显示出一定疗效,但效果有限;同时对于急性发作患儿,消除诱因和促进尽快入睡可很好控制发作 [24]。本研究发现了 4 个新的 ATP1A2/ATP1A3 基因变异位点,进一步加强了对 ATP1A2/ATP1A3 基因变异位点及相关临床表现的认识,为 ATP1A2/ATP1A3 基因变异相关疾病的靶向药物开发提供了理论依据。对于 ATP1A2/ATP1A3 基因变异相关疾病的鉴别要点见表 4,患儿如果能得到早期明确诊断,针对性治疗可改善其预后。
综上,本研究扩大了 ATP1A2/ATP1A3 基因变异相关疾病的临床表现谱和基因谱,提示在临床上,对于以癫痫、肌无力及偏瘫等神经症状为首发表现的疾病,特别是发病原因不明且常规治疗效果欠佳的疾病,应及时完善基因检测,明确诊断。
好文章,需要你的鼓励