




该病例为双罕见免疫疾病叠加、多重基因变异协同致病,存在常规治疗耐药、病情反复高热等多重诊疗难题。
团队依托精准基因检测与个体化靶向诊疗方案,明确致病根源并规范干预,助力患者病情好转、
为同类疑难罕见免疫重叠疾病的临床诊疗积累了宝贵经验。

患者为52岁男性,长期受多重慢性疾病困扰,病情迁延难愈、持续进展,诊疗难度极大。
患者拥有10年特发性血小板减少性紫癜病史,长期血小板数值偏低,存在潜在出血风险,既往未接受系统规范治疗;同时合并8年克罗恩病史,反复出现腹痛、腹泻症状,饮食刺激后症状加剧,肠道溃疡反复发作,病情始终无法有效控制。
入院前8个月,患者病情突发危重进展,出现持续性高热,最高体温达40℃,短期内体重骤降7kg,接受抗生素治疗后无明显效果,病情持续恶化。
多年来,患者辗转接受常规治疗,存在激素短期起效、减量后复发,英夫利昔单抗治疗耐药等问题,后续还出现全血细胞减少情况,最终陷入激素依赖、生物制剂耐药、血小板顽固性减少的三重临床难题。
针对该患者复杂危重的病情,医院立即启动疑难病例多学科会诊。同济大学附属第十人民医院刘占举教授团队参与诊疗指导,结合患者病史、治疗史及临床症状,判断患者并非简单的两种疾病叠加,存在特殊的内在致病机制。
诊疗团队逐一排查淋巴瘤、克隆性淋巴增殖性疾病等相似疑难病症,通过系统检查明确患者存在贫血、炎症指标显著升高等表现,影像学及内镜检查提示中重度活动性克罗恩病,骨髓检查证实造血异常、血小板减少,最终确诊为克罗恩病合并特发性血小板减少性紫癜重叠病症。

为从根源破解病情难治、反复、耐药问题,团队进一步完善基因检测,最终锁定核心病因。检测结果显示:患者存在IL10RA基因突变+1q21.1q32.1染色体片段重复复合型基因变异,属于罕见多重基因异常。
其中,IL10RA突变会损伤肠道抗炎机制,诱发顽固性肠道炎症;1q21.1q32.1染色体重复可引发造血异常,造成血小板持续减少。
两类基因异常协同作用,同时诱发肠道炎症、机体免疫紊乱与血液系统异常,也是患者常规治疗失效、病情持续进展的核心原因。
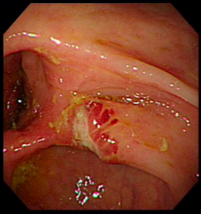
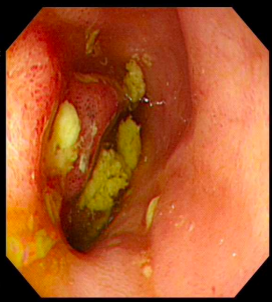
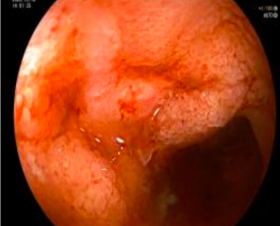
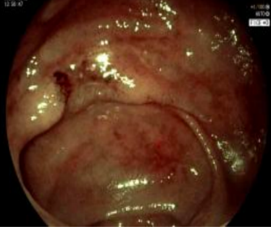
明确基因致病机制与病情特点后,团队为患者制定精准靶向治疗+激素阶梯减量的个体化诊疗方案,精准阻断机体异常致病通路,同步调控肠道炎症与免疫紊乱问题,适配该基因驱动型疑难病例的诊疗需求。

经过系统规范治疗,患者病情得到有效控制。治疗15天后,患者持续高热症状完全消退,各类不适症状显著缓解。
血小板数值从54×10⁹/L回升至正常水平(101×10⁹/L),体内炎症指标大幅回落,肠道活动性炎症得到有效控制,患者身体状态稳步恢复,达到康复出院标准。
据临床资料显示,克罗恩病合并特发性血小板减少性紫癜本身临床发病率极低,而本次病例由多重基因变异驱动、且对常规激素、生物制剂全面耐药,属于国内外少见的疑难免疫重叠病例,具备极高的临床研究与参考价值。
针对炎症性肠病患者,若出现不明原因反复高热、顽固性血小板减少、常规激素及生物制剂治疗耐药等情况,需高度警惕合并免疫性血液病的可能,及时完善骨髓检查、流式检测及基因检测,精准明确病因。
好文章,需要你的鼓励