



来源:《临床儿科杂志》
张未, 汪洋, 邓文华, 吴亚斌. 14 例原发性纤毛运动障碍临床表现、纤毛结构及基因特点分析 [J]. 临床儿科杂志, 2025, 43(9): 680-685 DOI:10.12372/jcp.2025.25e0254
ZHANG Wei, WANG Yang, DENG Wenhua, WU Yabin. Analysis of clinical manifestations, ciliary structure and genetic characteristics of primary ciliary dyskinesia in 14 children[J]. Journal of Clinical Pediatrics, 2025, 43(9): 680-685 DOI:10.12372/jcp. 2025.25e0254
本文作者: 张 未 汪 洋 邓文华 吴亚斌
作者单位: 湖北省妇幼保健院(湖北武汉 430070)
摘要:目的 探讨原发性纤毛运动障碍(PCD)临床表现、纤毛结构及基因变异特点。方法 回顾性分析 2017 年 1 月至 2024 年 12 月在医院经支气管镜黏膜活检电镜检查或二代测序全外显子组基因检测确诊为 PCD 患儿的临床资料。结果 共纳入 14 例患儿,男 4 例、女 10 例,中位发病年龄 6.3(1.3~7.9)岁,中位诊断年龄 8.0(7.0~10.8)岁。主要临床表现包括慢性湿性咳嗽(14 例,100%),鼻/鼻窦炎(12 例,85.7%),支气管扩张及肺不张(9 例,64.3%),内脏转位(3 例,21.4%),慢性中耳炎(2 例,14.3%)。14 例患儿支气管镜下均表现为化脓性支气管炎(100%),2 例镜下合并支气管狭窄,1 例合并支气管闭塞。10 例患儿在急性感染控制至少 2 周后检测鼻呼出一氧化氮(nNO),8 例 nNO 均明显降低,2 例 nNO>77 nL/min。7 例患儿进行全外显子组检测,其中 2 例阴性,5 例检出双等位基因变异,DNAH5 基因 2 例,CCNO 基因 2 例,DNAH1 基因 1 例。结论 PCD 临床表型以慢性咳嗽、鼻窦炎、支气管扩张为主,内脏转位及新生儿期呼吸窘迫发生率低,部分患儿 nNO 高于阳性阈值,并发支气管闭塞,主要基因变异为 DNAH5 和 CCNO。
关键词:原发性纤毛运动障碍;纤毛超微结构; 基因变异;儿童
原发性纤毛运动障碍(primary ciliary dyskinesia,PCD)是纤毛结构缺陷和/或运动障碍造成纤毛黏液清除能力降低或丧失的疾病,是一种罕见的遗传异质性疾病,可导致新生儿呼吸窘迫,慢性气道感染,器官偏侧发育异常,不孕不育等 [1-2]。40%~55% 的 PCD 患者存在内脏反位,表现为支气管扩张、内脏转位、鼻窦炎三联征,称为 Kartagener 综合征 [1]。目前已报道 50 多个 PCD 相关的致病基因 [3],其遗传模式主要为常染色体隐性遗传,另有 X 连锁隐性及常染色体显性遗传,不同国家人群 PCD 遗传谱存在差异,最常报道的基因型是 DNAH5、DNAH11、CCDC40 等 [4]。随着鼻呼出一氧化氮(nasal nitric oxide,nNO)、透射电子显微镜(transmission electron microscopy,TEM)纤毛超微结构检查的推广及全外显子组基因检测技术的应用,为 PCD 患者的诊断提供了帮助。为提高该疾病的诊治水平,本研究总结湖北省妇幼保健院 2017 年 1 月至 2024 年 12 月经支气管黏膜活检纤毛电镜检查、基因检测确诊的 14 例 PCD 患儿的临床资料,分析其临床特点、影像学表现、纤毛结构异常及基因变异特点。
1 对象与方法
1.1 研究对象
回顾性分析 2017 年 1 月至 2024 年 12 月在湖北省妇幼保健院诊断为 PCD 患儿的临床资料。纳入标准:(1)起病年龄:0~18 岁;(2)儿童 PCD 诊断参照《原发性纤毛运动障碍诊断与治疗中国专家共识》[1]。符合 PCD 临床特征(①足月婴儿不明原因的新生儿呼吸窘迫,②半岁前开始的全年每日咳嗽,③半岁前开始的全年持续性非季节性鼻窦炎,④内脏反位)中至少 2 项,再结合以下任意 1 项阳性结果(①公认的纤毛超微结构缺陷,②PCD 相关基因的双等位基因致病性突变),可确诊为 PCD。例 6 患儿为 Kartagener 综合征,即使未完全满足上述标准,也被认为是 PCD。排除标准:①临床资料不完整;②免疫缺陷、囊性纤维化、先天性气道畸形等疾病。
1.2 方法
1.2.1 鼻呼出一氧化氮测定 使用 Sunvou-CA2122®(尚沃医疗,中国)一氧化氮装置测量 nNO,nNO 流速为 10 mL/s。在急性感染控制至少 2 周后,对于>5 岁的配合检查的儿童进行 nNO 测量,通过将 nNO 浓度(ppb)乘以采样流速(0.6L/min)来计算 nNO 值(以 nL/min 为单位)。
1.2.2 透射电子显微镜检查 在急性感染控制至少 8 周后,10 例患儿进行支气管镜检查并行支气管黏膜活检,活检部位为右中叶开口分叉处,右下叶基底干与背段分叉处,左上下叶开口分叉处,获取黏膜标本后立即放入 2% 戊二醛标本固定液,送至武汉大学湖北省人民医院电镜病理中心行 TEM 检查。
1.2.3 基因检测 征得家属知情同意后,7 例患儿行基因检查。取患儿及父母外周血 2 mL,5 例(例 1、2、3、6、11)送至北京智因东方转化医学研究中心有限公司,例 4 送福君基因(福州福瑞医学检验实验室),例 5 送厦门基源医学检验实验室,提取基因组外周血 DNA,用二代测序进行全外显子组家系(trio WES)检测,根据美国医学遗传学与基因组学学会(ACMG)指南对变异位点进行致病性评级,阳性变异位点采用 Sanger 测序验证。
1.3 统计学分析
采用 SPSS 20.0 进行数据分析。非正态分布的计量资料以 M(P25~P75)表示。计数资料以 n(%)表示。
2 结果
2.1 临床特征
14 例患儿中,男 4 例、女 10 例,中位发病年龄 6.3(1.3~7.9)岁,中位诊断年龄 8.0(7.0 ~10.8)岁。例 6 为 Kartagener 综合征。
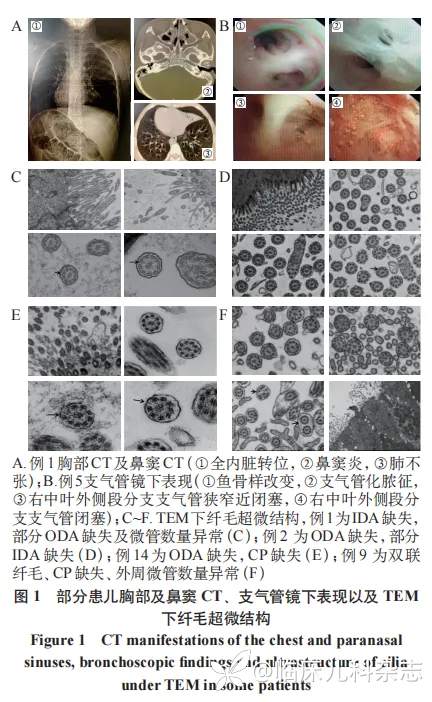
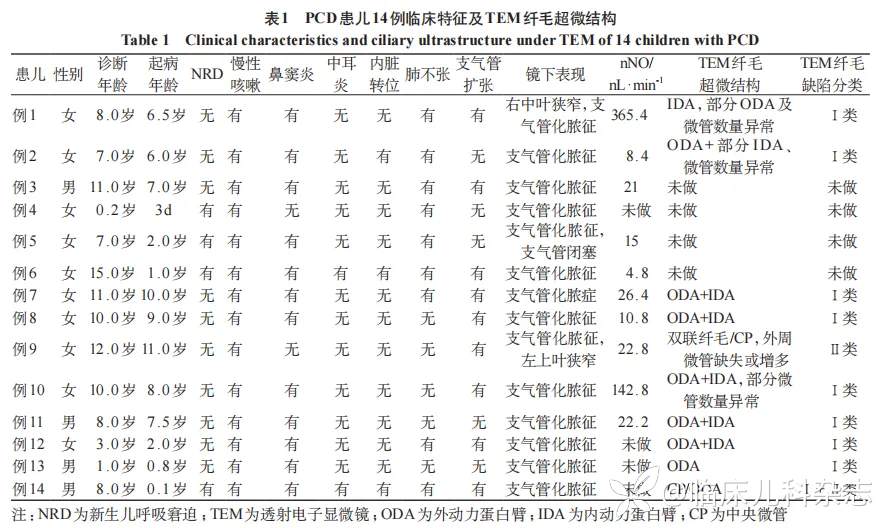
14 例(100%)患儿均有慢性湿性咳嗽,鼻/鼻窦炎 12 例(85.7%),支气管扩张 9 例(64.3%),急性期伴有肺不张 9 例(64.3%),慢性中耳炎 2 例(14.3%),新生儿期有急性呼吸窘迫综合征(RDS)4 例(28.6%),内脏转位 3 例(21.4%)。见图 1。14 例患儿支气管镜下表现均为化脓性支气管炎,2 例(14.3%)合并局部支气管狭窄,1 例(7.1%)伴发支气管闭塞等,见表 1、图 1。
2.2 鼻呼气一氧化氮测定结果
10 例患儿在急性感染控制至少 2 周后完成 nNO 检查,其中 8 例(80%)nNO 值小于阳性阈值(77 nL/min),2 例(20%)nNO 值正常。见表 1。
2.3 透射电子显微镜结果
10 例患儿纤毛 TEM 检查均为阳性,其中内动力蛋白臂(IDA)缺失+外动力蛋白臂(ODA)缺失 8 例,中央微管(CP)缺失 2 例、双联纤毛 1 例,微管数量异常 4 例,部分患儿多种结构异常同时存在(见表 1、图 1)。
2.4 基因检测结果
7 例患儿(例 1~例 6,例 11)进行全外显子组基因检测,其中 2 例检测结果为阴性 [例 6 仅检测到 DNAH5 基因 c.8569 G>T(NM_001369)单杂合变异;例 11 未发现可疑变异],5 例(例 1 ~例 5)检出双等位基因异常;共检测到 3 个基因上 9 个点变异,DNAH5 基因 2 例,CCNO 基因 2 例,DNAH1 基因 1 例。见表 2。
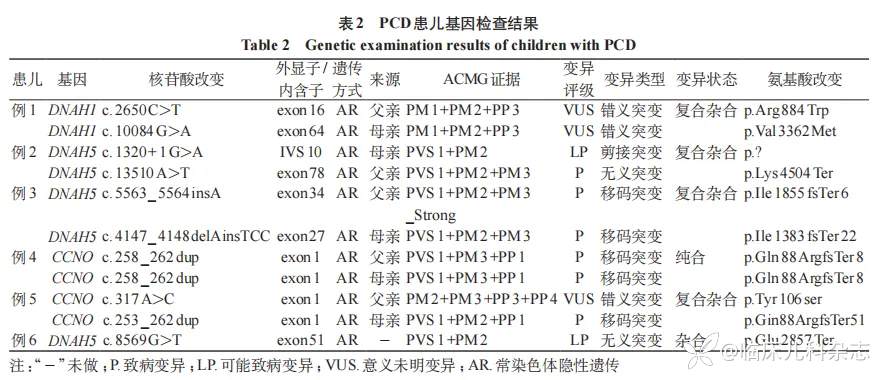
例 1 携带 DNAH1 基因 c.2650C>T 和 c.10084 G>A 的复合杂合突变,c.2650C>T 错义突变来源于父亲,导致氨基酸改变:p.R884W;c.10084 G>A 错义突变来源于母亲,导致氨基酸改变:p.V3362M。例 2 携带 DNAH5 基因 c.1320+1 G>A 和 c.13510A>T 复合杂合突变,c.1320+1 G>A 剪接突变来源于母亲,c.13510A>T 无义突变来源于父亲,导致氨基酸编码终止:p.K4504*。例 3 同时检出 DNAH5 基因变异,DNAH5 基因的 c.5563_5564ins A 和 c.4147_4148delA insTCC 复合杂合突变分别来源于父亲和母亲,导致氨基酸发生移码提前终止 :p.I1855Nfs*6 和 p.I1383Sfs*22。
例 4 及例 5 均为 CCNO 基因异常。例 4 携带 CCNO 基因 c.258_262dup 纯合移码突变,父母杂合携带,导致氨基酸移码提前终止:p.Gln88Argfs*8。例 5 携带 CCNO 基因的 c.317A>C 和 c.253_262dup 的复合杂合突变,c.317A>C 错义突变来源于父亲,导致氨基酸发生替换:p.Tyr106ser;c.253_262 dup 移码突变来源于母亲,导致氨基酸移码提前终止 :p.Gln88Argfs*51。c.317A>C 及 c.253_262dup 暂未被 ClinVar、mastermind 数据库收录,c. 253_262 dup 基因位点依据 ACMG 指南评为致病性变异(PVS1+PM2+PP1),已被 Guan 等 [5] 报道。c.317A>C 基因位点依据 ACMG 指南评为意义未明的变异(PM2+PM3+PP3+PP4),暂无文献报道。
3 讨论
本研究纳入的 14 例 PCD 患儿中位发病年龄 6.3 岁,明显大于 Guan[5] 等报道的 3 个月,可能与既往对 PCD 的认知不足,家长关注度不够,回顾既往病史偏差等因素有关。中位诊断年龄为 8.0 岁,大于欧洲的 5.3 岁和美国的 3 岁 [6-7]。可能与既往对 PCD 疾病认知及检测手段有限导致诊断延误有关。本研究观察到典型的 PCD 临床特征,包括慢性湿性咳嗽(100%)、鼻/鼻窦炎(85.7%)、支气管扩张(64.3%)、新生儿呼吸窘迫综合征(NRDS,28.6%)、内脏转位(21.4%)和慢性中耳炎(14.3%),与既往研究结果 [8] 一致。其中内脏转位发生率 21.4%,低于既往报道的 42%[4],Raidt 等 [4] 研究显示各区域内脏转位发生率分别为 28%(土耳其)、31%(荷兰)、37%(德国)、40%(以色列),提示内脏转位发生率在不同国家之间差异较大。本研究中 NRDS 发生率较低,国外有研究报道其发生率约 55%[9],甚至高达 80%[10],故当足月新生儿出现不明原因的呼吸窘迫时,应考虑本病的可能,及时识别并完善检查,以早期诊断及干预 [11]。本研究 14 例 PCD 患儿支气管镜下表现均为化脓性支气管炎,考虑与纤毛运动及功能降低,呼吸道分泌物聚集相关;2 例患儿支气管镜下表现为局限性支气管狭窄,考虑与炎症反应、局部肿胀增生相关;1 例患儿合并右中叶外侧段支气管闭塞,临床表现为右中叶局限性肺不张、毁损肺。
欧洲呼吸学会和美国胸科学会均建议使用 nNO 检测作为具有典型 PCD 病史患者的主要筛选工具 [12]。有研究表明,以<77 nL/min 为 nNO 阳性阈值时,诊断 PCD 的灵敏度为 98%,特异度为 99%[13]。本研究中 10 例患儿进行 nNO 检测,80% 患儿的 nNO 值< 77nL/min。PCD 患者中 nNO 值较低的原因包括 nNO 代谢增加、气道上皮细胞产生 NO 减少、鼻窦异常导致 NO 阻塞或产生 NO 减少等 [14]。既往研究发现部分 PCD 患者的 nNO 值>77 nL/min,相关基因变异包括 RSPH1、FOXJ1、CCNO、GAS8 等 [3]。本研究中 2 例患儿 nNO 高于阳性阈值,其中 1 例变异位于 DNAH1 基因。PCD 患儿 nNO 高于阈值的原因尚不明确,相关研究发现高 nNO 值不仅与特定基因(RSPH 1、TTC12、FOXJ1 等)有关,还与其他基因的潜在亚型突变有关(可能产生功能性蛋白质)[15]。有研究认为没有纤毛超微结构缺陷的 PCD 患者似乎有更高的 nNO 值,容易延误 PCD 的诊断 [16]。
2020 年发布的 TEM 国际共识指南 [17] 确定了 PCD 纤毛超微结构诊断的两类缺陷:Ⅰ类缺陷,包括 ODA 缺失、ODA+IDA 缺失、IDA 缺失+微管紊乱,结合典型临床表现可确诊 ;Ⅱ类缺陷,包括中央微管缺失、纤毛数量少、微管紊乱等,确诊需进一步结合其他诊断手段。文献报道发现纤毛的超微结构与基因有较好的相关性,DNAH5 基因变异表现为 ODA 缺失,DNAH1 基因编码的纤毛超微结构正常 [14]。本研究例 1 患儿为 DNAH1 基因变异,TEM 结果显示 IDA 缺失,部分 ODA 缺失伴有微管数量异常,与文献报道不一致,考虑与反复慢性感染或气道炎症导致纤毛超微结构异常相关。研究发现约 30% 的 PCD 患者仅有纤毛功能异常,而纤毛结构正常 [18],因此,单独的 TEM 正常不能排除 PCD,需要行高速视频成像分析、免疫荧光分析法或基因检测等诊断测试进一步明确诊断。
目前已发现>50 个与 PCD 相关的基因 [14],既往研究发现 [19],PCD 常见基因型存在种族及地域差异,DNAH11 和 DNAH5 在欧洲、东亚人群最为常见;南亚人群以 DNAH11 和 ODAD2 为主,而在芬兰和非洲人中,DNAH11 和 DNAI1 更常见。本研究共检出 2 例 DNAH5、2 例 CCNO 及 1 例 DNAH1,与上述结果相似。Benkhelifa[20] 等认为 DNAH1 在睾丸中表达显著高于气管细胞,报道病例仅表现为男性弱精,而无 PCD 其他系统表现;而 Imtiaz[21] 等报道的 DNAH1 变异患者有慢性鼻窦炎、支气管扩张及内脏转位等典型 PCD 表现。本研究中例 1 患儿仅表现为慢性湿性咳嗽、反复肺炎表现,无内脏转位等,表明相同基因型仍具有明显异质性。CCNO 基因变异导致多运动纤毛的生成减少,与 PCD 的其他遗传原因相比,可导致更严重的气道疾病 [22]。有研究认为 CCNO 变异患儿出现症状较早,89% 为新生儿发病,61.8% 患有 NRDS[23],且均未观察到内脏转位 [24]。本研究中 2 例 CCNO 变异患儿均为幼年起病,其中例 4 生后 3 天出现呼吸困难症状,合并 NRDS,2 例患儿均无内脏转位,与文献报道 [24] 一致。本研究中例 4 携带 CCNO 基因 c.258_262dup 纯合突变,该位点在目前已鉴定出的 CCNO 基因变异中最为常见;例 5 为 CCNO 基因复合杂合致病性变异,c.317 A>C 及 c.253_262dup 突变位点暂未被 ClinVar、mastermind 数据库收录,Guan[5] 等曾报道 c. 253_262dup 突变位点,c.317A>C 突变位点暂无相关文献报道,考虑 c.317A>C 为 CCNO 新发现的变异位点。
总之,本研究总结了 PCD 患儿的临床表现、纤毛表型和遗传谱多样性,以期提高临床医师对此种疾病异质性的认知度,有助于更好诊断 PCD。对幼年起病,反复呼吸道感染,合并鼻窦炎或中耳炎、支气管扩张患儿,均需警惕 PCD 的可能,临床病史分析结合 nNO 筛查、TEM 检查和/或基因检测是确诊 PCD 的主要手段。治疗方面,应实施气道廓清、及时抗感染治疗,耳鼻咽喉科与呼吸科应积极开展多学科合作,共同管理患儿的上、下呼吸道问题;而规律随访对预防 PCD 患儿严重并发症,改善其生存质量十分重要 [25]。对存在不孕/不育的 PCD 患者,可采取辅助生殖技术治疗。目前基因治疗是 PCD 患儿的精准化治疗,如基因特异性 mRNA 替代,基因编辑等仍在研究中 [26]。
好文章,需要你的鼓励