



来源:《临床儿科杂志》
李苗苗, 潘桂梅, 刘磊, 陈琼, 李杨世玉, 张子夏, 王曦, 杜萌萌, 卫海燕, 陈永兴. 甲状腺结合球蛋白缺乏症 3 个家系报告 [J]. 临床儿科杂志, 2025, 43(8): 598-603 DOI:10.12372/jcp.2025.25e0349
LI Miaomiao, PAN Guimei, LIU Lei, CHEN Qiong, LI Yangshiyu, ZHANG Zixia, WANG Xi, DU Mengmeng, WEI Haiyan, CHEN Yongxing. Thyroxine binding globulin deficiency in three families and review of the literature[J]. Journal of Clinical Pediatrics, 2025, 43(8): 598-603 DOI:10.12372/jcp.2025.25e0349
本文作者: 李苗苗 1 潘桂梅 2 刘 磊 3 陈 琼 1 李杨世玉 1 张子夏 1 王 曦 1 杜萌萌 1 卫海燕 1 陈永兴 1
作者单位:1. 郑州大学附属儿童医院 河南省儿童医院 郑州儿童医院内分泌遗传代谢科(河南郑州 450053);2. 青岛大学附属妇女儿童医院儿童重症监护室(山东青岛 266011);3. 郑州大学附属儿童医院 河南省儿童医院 郑州儿童医院儿研所 河南省儿童遗传代谢性疾病重点实验室(河南郑州 450018)
摘要:目的 旨在探讨甲状腺结合球蛋白(TBG)缺乏症的临床特征和遗传学特点,提升临床诊断准确性,避免误诊及过度治疗。方法 对来自 3 个不同家系的成员进行 SERPINA7 基因测序,探究其遗传基础。结果 通过全外显子组测序检测出 3 个家系的变异均发生在外显子,分别为 c.712A>G(p.M238V)、c.1114delC(p.L372Ffs*23)、c.383-401dup(p.F135Afs*21),其中家系 1、家系 3 的变异属首次报道。结论 TBG 缺乏症无需特殊治疗,正确的诊断与全面的疾病教育是该病管理的关键。本研究发现了 2 个新的 SERPINA7 基因变异,拓展了该基因的变异谱。
关键词:TBG 缺乏症;SERPINA7;基因检测
先天性因素引起的甲状腺结合球蛋白缺乏症(thyroxine binding globulin deficiency,TBGD)是一种 X 连锁遗传性疾病 [1],患病率为 1/7000 至 1/3000,男女比例为 8.9∶1[2]。该疾病是由 SERPINA7 基因变异导致 TBG 水平下降,引起三碘甲状腺原氨酸(TT3)和甲状腺素(TT4)水平下降,而游离甲状腺素(FT4)、游离三碘甲状原氨酸(FT3)正常,这种甲状腺功能指标的「分离」现象是 TBG 缺乏症的重要特征。根据 TBG 缺乏的程度,可以分为完全性缺乏(TBG-CD)、部分性缺乏(TBG-PD)。在国内临床实践中,由于对该病的认知不足,误诊情况时有发生。本研究回顾性分析了 3 例 TBG-CD 患儿及其家系的临床资料,为临床医师提供更为精准的诊断思路,有效减少误诊误治现象的发生。
1 对象与方法
1.1 研究对象
回顾性分析 2019 年 10 月至 2025 年 2 月就诊于郑州大学附属儿童医院的 3 例 TBG-CD 患儿的临床资料。纳入标准:①甲状腺功能检测提示:TT3、TT4 水平降低,FT3、FT4 正常;②基因检测结果提示 SERPINA7 基因存在变异;③TBG<3.50 µg/mL。排除标准:临床资料不完整者。
1.2 方法
1.2.1 临床资料 通过电子病历系统采集 3 例患儿的临床资料,包括性别、年龄、既往史、家族史、体格检查、实验室检查、影像学检查、治疗及随访情况等。其中实验室检查主要包括甲状腺功能检测、TBG 检测及基因检测等,影像学检查包括骨龄检查、甲状腺超声、垂体磁共振成像(MRI)等。
1.2.2 基因检测 在获得患儿及家长知情同意后,采集患儿及其父母的外周血各 2 mL,乙二胺四乙酸(EDTA)抗凝,由第三方基因检测机构对患儿及其父母的血液样本进行全外显子测序(WES)检测。检测区域包括人类全外显子组中 20099 个基因的外显子区域及旁侧 20bp 内的内含子区域。同时,采用 Sanger 测序法对筛选出的变异位点进行家系验证。
2 结果
2.1 一般临床资料
来自 3 个家系的 3 例患儿就诊年龄分别为 3 岁 6 个月、8 岁 9 个月、5 岁 1 个月,3 例患儿中 1 例为女性,2 例为男性。3 例患儿分别以「生长缓慢 3 年余」「发现甲状腺功能异常 7 年」「生长迟缓 2 年」为主诉入院治疗。例 1 和 2 曾被误诊为甲状腺功能减退症并分别给予甲状腺素 25~50 μg/d 治疗 5 月 13 天、6 年 7 月。例 1 的体格检查可见颈蹼、双手通贯掌,例 2 入院前近 3 个月出现了注意力不集中、易急躁的表现。
2.2 实验室检查
3 例患儿的肝肾功能及心肌酶检查未见异常,甲状腺功能五项结果均提示 TT3 及 TT4 降低、FT4 及 FT3 正常、TSH 正常或降低,TBG 水平均<3.50 μg/mL。例 2 的抗甲状腺球蛋白抗体、甲状腺过氧化物酶抗体、促甲状腺素受体抗体均处于正常参考范围。
影像学检查提示:例 1 根据 Greulich-Pyle (GP) 图谱判断骨龄为 3 岁,心电图提示窦性心律不齐;甲状腺超声未见异常。例 2 根据 GP 图谱法判断患儿的骨龄为 10 岁,大于实际年龄;心电图提示窦性心动过速;甲状腺超声提示甲状腺体积稍小;垂体 MRI 示垂体高约 2.0 mm。例 3 根据 GP 图谱法判断骨龄为 4 岁 6 个月;心电图提示窦性心律不齐;垂体 MRI 示垂体高约 2.6 mm。
2.3 基因检测结果
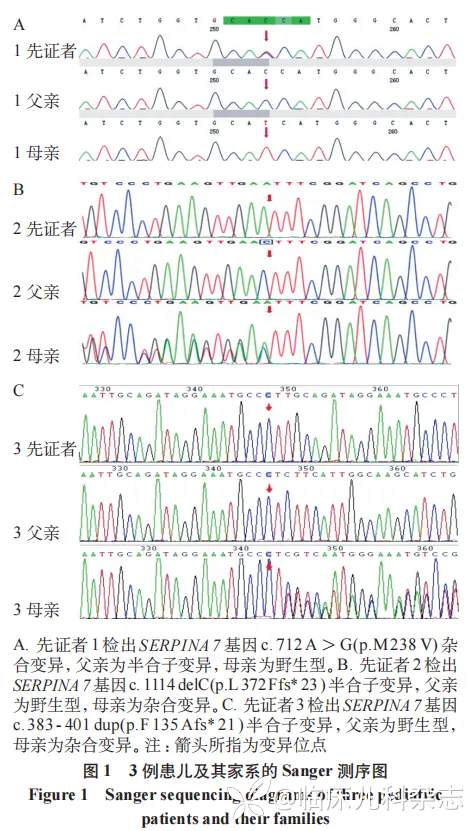
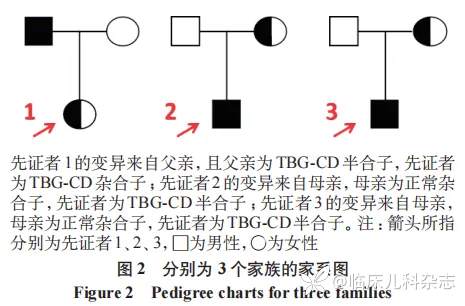
经过基因检测 ,在 3 例患儿中 ,分别发现 SERPINA7 基因 3 号外显子存在一杂合错义变异 :c.712A>G(p.M238V)、5 号外显子存在半合子移码变异:c.1114delC(p.L372Ffs*23)、2 号外显子存在半合子移码变异:c.383-401dup(p.F135Afs*21)。Sanger 测序结果分别提示变异源自父亲、母亲、母亲,具体 Sanger 测序结果见图 1,家系图见图 2。其中例 1 的父亲多次行甲状腺功能五项结果提示 TT3、TT4 均降低,TSH、FT3、FT4 均正常,TBG<3.50 µg/mL,最后也确诊为 TBG-CD。参照美国医学遗传学与基因组学学会(ACMG)指南,3 例患儿变异评级分别为意义不明变异、意义不明变异、可能致病性变异。
2.4 治疗与随访
例 1 既往甲状腺素治疗过程中多次随访复查甲状腺功能五项提示 TT3、TT4 较低,而 FT3、FT4 正常,初诊时建议停用甲状腺素,患儿现已停用甲状腺素治疗 4 年 8 个月余,随访过程中生长发育等未见异常。例 2 入院治疗时已行甲状腺素治疗 6 年 7 个月,出现注意力不集中、易急躁等高代谢症状,且骨龄提示大于实际年龄,明确诊断后立即停用甲状腺素,现患儿 8 岁 11 个月,停药后多次复诊无异常。例 3 初诊完善相关检查后即确诊,未进行甲状腺激素治疗,随访至今无异常表现。
3 讨论
TBG 是循环中甲状腺激素的主要载体,亲和力最高,约 70%~75% 的甲状腺激素与其结合,其余与甲状腺转运蛋白和人血清白蛋白结合 [3],其主要功能是维持血清甲状腺激素总量恒定,防止波动 [4]。故当 TBG 的数量或亲和力下降将会导致 TT3、TT4 水平的降低,但不影响 FT3、FT4 的水平,通常情况下也不会导致甲状腺功能减退的症状,不过少数 TBGD 患者可能合并甲状腺功能减退症 [5]、甲状腺功能亢进症 [6-7] 等其他甲状腺异常疾病,使化验结果和疾病鉴别难度增加 [4]。
在临床上,评估儿童甲状腺功能常用的检查组合是甲状腺功能三项,TBG 检测很少用于疾病诊断和治疗 [8],同时也缺乏针对 TBGD 的特异性诊断路径。本文例 1、2 因多次复查甲状腺功能仍提示 TT3、TT4 低,完善血清 FT3、FT4 及 TBG 后得到明确诊断。当儿童甲状腺功能检测提示 TT3、TT4 降低,但 TSH 正常,在考虑中枢性甲状腺功能减退的同时,也应高度怀疑 TBGD,尤其对于没有乏力、皮肤苍黄粗糙、便秘、心率减慢等甲状腺功能减退症临床表现的患儿,需进一步检测血清 FT3、FT4 及 TBG 水平。若血清 FT3、FT4 正常,血清 TBG 难以检测或水平极低(低于 5 mg/L 或 0.9nmol/L,或低于正常平均值的 0.003%),可诊断为 TBG-CD[8];若血清 TBG 水平有所降低但仍可检测到,则为 TBG-PD。在极少数情况下,SERPINA7 的变异会导致功能性 TBG 缺陷,使 TBG 与 T4 结合的亲和力下降,出现 TT4 降低但 TBG 水平正常的情况 [9],此时仅靠临床检验难区分,需尽早进行基因检测以明确是否携带相关基因变异,实现早期确诊。
对于因生长迟缓或身材矮小 [8] 检测甲状腺功能异常时,可能更易被误诊为中枢性甲状腺功能减退症。然而,这些患儿往往缺乏其他典型的甲状腺功能减退症临床表现,因此详尽的病史采集和细致的体格检查显得尤为重要。同时 , 要提高临床医师对 TBG 缺乏症这一疾病的认识, 及时发现甲状腺功能指标的「分离」现象,通过检测 TBG 水平、SERPINA7 基因变异分析明确 TBG 缺乏症诊断。若误诊为甲状腺功能减退症并给予多余甲状腺素治疗,就极有可能出现甲状腺毒症等不良反应,诸如心悸、多汗、体重减轻、骨质疏松、骨龄进展快等,尤其在妊娠期妇女和青少年儿童中 [10]。本研究中,例 1、2 初诊时因生长迟缓且 TT3、TT4 降低,被误诊为甲状腺功能减退症并给予甲状腺素治疗,例 2 出现了甲状腺素过量使用的不良反应。
TBG 是由 SERPINA7 基因编码并在肝脏合成的一种 54KDa 糖蛋白 [11],属于丝氨酸蛋白酶抑制剂超家族成员。SERPINA7 基因位于 X 染色体长臂 q22.2 区域,包含 5 个外显子,编码的 TBG 前体含 415 个氨基酸残基,成熟分子含 395 个氨基酸残基 [12]。对已报道该病的 SERPINA7 基因变异类型进行分析发现 [5,8-9,11,13-24],目前已知有 51 种 SERPINA7 基因的变异,约 58.9%(29/51)为错义或无义变异,约 9.8%(5/51)为剪切变异,多为单核苷酸的替换或缺失,常见于外显子 5 和外显子 4 上(具体变异分布及比例见图 3),另有文献报道 [17]4 个家系的 TBG 缺乏症是由 SERPINA7 基因下游 20kb 处增强子单碱基变化引起。最常见的热点变异为 p.Leu372Phefs*23,也称为日本型完全缺乏(TBG-CDJ),其在亚洲人群中较为常见 [22]。既往文献还表示移码变异或无义变异更易引起 TBG-CD,而错义变异更易引起 TBG-PD,而男性更易患 TBG-CD[25]。本研究中 TBG-CD 的 3 种变异均发生在外显子,2 种属首次报道,这拓宽了 SERPINA7 基因的变异谱。家系 1 所携带错义变异 c.712A>G(p.M238V),位于第 3 外显子,使得蛋白第 238 位的甲硫氨酸(Met)变成了缬氨酸(Val)。家系 2 所携带外显子 5 上的移码变异 c.1114delC(p.L372Ffs*23)和家系 3 所携带外显子 2 上的移码变异 c.383 -401 dup(p.F135Afs*21 ) 均导致蛋白质的翻译和合成提前终止。本研究中例 1、2 的变异被评定为意义不明的变异,尚需进一步行细胞或动物功能研究来验证这些变异的影响。
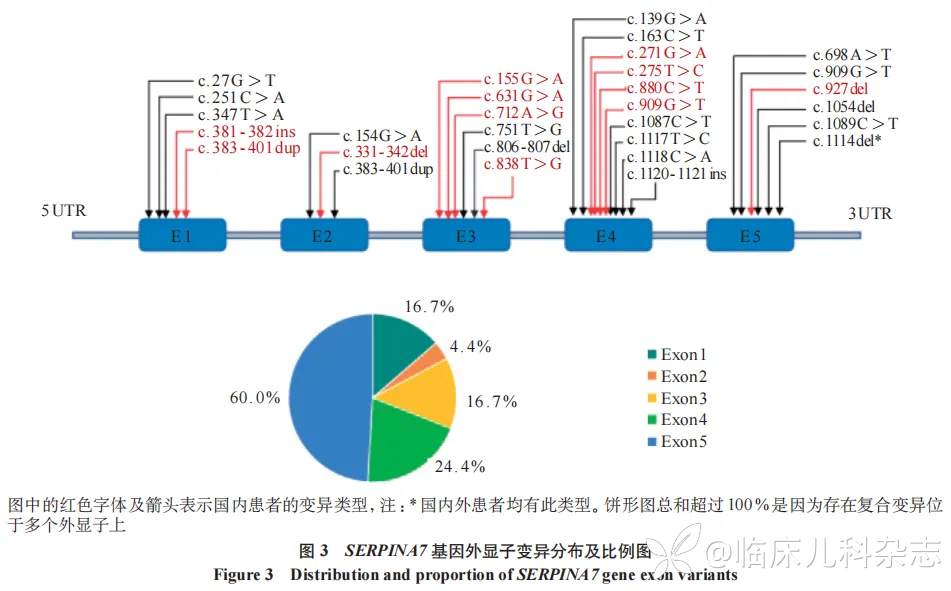
该病属于 X 连锁遗传,理论上讲女性杂合携带者不致病,但回顾既往文献发现已报道了 TBG-CD 女性患者 9 例、TBG-PD 女性患者 14 例。在本研究中,例 1 为 TBG-CD 杂合子女童,其 TBG 水平低于 3.50 µg/ml,与半合子男童例 2、3 的 TBG 水平相当。女性杂合携带者出现 TBG 缺乏症可能涉及两种机制。一种是一条 X 染色体 SERPINA7 基因存在变异,另一条 X 染色体完全缺失,这时候患儿表现为 TBG 缺乏症合并特纳综合征,既往文献中也曾报道过两例 45,XO 核型的特纳综合征女性存在 TBG-CD 表型 [25-26]。这时临床医师不仅要关注 TBG 缺乏相关的表现,还应留意特纳综合征的临床体征,如身材矮小、颈蹼、盾形胸、乳头间距增宽等,提示医师应该进一步完善染色体核型分析等检查,避免漏诊。另一种可能的机制是一条 X 染色体 SERPINA7 基因变异,另一条 X 染色体选择性偏移失活,这时需要关注患者是否存在其他与 X 染色体连锁基因相关的异常表现。例 1 尽管有颈蹼、生长缓慢,但无其他异常体征,基因检测也未发现 X 染色体拷贝数异常,因此考虑可能为 X 染色体失活偏移 [27],未来仍需要我们进一步随访观察,监测其生长发育、内分泌功能等相关表现,深入探索 X 染色体失活偏移与 TBGD 的关联机制,为临床诊疗与遗传咨询提供更充分的依据。
该病是预后良好的良性疾病,管理该病的关键环节在于实现正确的诊断以及全面的疾病教育。正确的诊断可避免误诊导致的过度医疗行为,不仅能降低患者家庭额外的经济开支,还能有效地减轻家属在心理和经济方面承受的压力。疾病教育方面,临床医师应向患者及其家属详细介绍 TBGD 的发病机制、遗传模式、临床特征及与其他甲状腺疾病的区别,重点强调该病虽会导致甲状腺功能指标异常,但通常不会对身体健康造成实质性损害。同时告知家属无需因该病对患儿生活进行过度限制,有效消除家属因疾病认知不足引发的恐惧与焦虑情绪。
综上所述,先天性 TBGD 无特异性表现,当儿童甲状腺功能检测时发现 TT3、TT4 降低而 TSH 正常,但缺乏甲状腺功能减退症的典型临床表现,或甲状腺功能检查出现「分离」现象时,应该留意该病,进一步检测血 TBG 水平,必要时完善基因检测以鉴别甲状腺功能减退症与 TBGD,避免误诊和甲状腺素的过度替代治疗。该症无需特殊治疗,正确的诊断与全面的疾病教育才是该病管理的关键。本研究发现了 2 个新的 SERPINA7 基因变异,拓展了该基因的变异谱。
好文章,需要你的鼓励